Paweł Z. Hrycaj, Jan K. Łącki
Od zwyrodnienia do zapalenia – współczesne poglądy na patogenezę choroby zwyrodnieniowej stawów
From osteoarthrosis to osteoarthritis – current view on the pathogenesis of degenerative joint disease
z Kliniki Reumatologii i Immunologii Klinicznej Akademii Medycznej
im. Karola Marcinkiewicza w Poznaniu
Kierownik Kliniki: dr hab. med. Jan K. Łącki
Streszczenie
Summary
Osteoarthritis is one of the most common rheumatic disorders and poses an important sociomedical challenge. Recently, there has been an increasing interest in the osteoarthritis resulting from the change in the understanding of the pathogenesis of the disease. For many years degenerative joint disease was regarded as a simple, age related, „wear and tear” problem. However, recent studies provide evidence that osteoarthritis results from a complex interplay of genetic, biologic, biomechanic, metabolic, inflammatory and imunologic factors. A number of mediators including cytokines operate in osteoarthritis and regulate the cartilage metabolism, activate matrix metalloproteases and the other proteolytic enzymes, and participate in the formation of osteophytes. The understanding of the pathomechanisms of osteoarthritis has important practical implications – it promotes research in the area of the novel treatment options like cytokine inhibitors, protease and nitric oxide inhibitors, stimulators of the cartilage matrix synthesis and chondrocyte growth, and the gene therapy.

WSTĘP
Zapewne trudno znaleźć lekarza, który nie zetknąłby się chociaż z jednym chorym na chorobę zwyrodnieniową stawów. Choroba ta jest bowiem powszechna i dotyczy znacznego odsetka populacji, zwłaszcza osób starszych. Jeszcze do niedawna uważana za objaw starzenia się i prostego „zużycia” chrząstki stawowej, od kilku lat znalazła się ponownie w centrum zainteresowania badaczy i lekarzy. „Osteoarthritis: time to shift the paradigm” – tak zatytułował swój opublikowany w 1999 roku w British Medical Journal artykuł redakcyjny wybitny znawca choroby zwyrodnieniowej, prof. Paul Dieppe (1). W rzeczy samej, na przestrzeni ostatnich lat zaszły duże zmiany w rozumieniu istoty i znaczenia choroby zwyrodnieniowej stawów. Z klinicznego punktu widzenia konieczne jest lepsze zbadanie roli czynników ryzyka i ustalenie kryteriów prognostycznych choroby, ustalenie standardów leczenia dla różnych postaci klinicznych choroby, weryfikacja skuteczności i wskazań do stosowania nowych leków, w tym tzw. leków określanych jako modyfikujące przebieg choroby. Postęp w leczeniu choroby zwyrodnieniowej stawów nie jest jednak możliwy bez gruntownego poznania jej patomechanizmu. Dziś wiemy, że przyczyny choroby zwyrodnieniowej są złożone: w powstawaniu i w rozwoju zmian stawowych uczestniczą różnorodne czynniki – biologiczne, biomechaniczne, metaboliczne, zapalne, immunologiczne, a ich względny udział w rozwoju choroby u poszczególnych chorych może być znacząco różny.
ZMIANY STAWOWE W PRZEBIEGU CHOROBY ZWYRODNIENIOWEJ
Zmiany zwyrodnieniowe dotyczą wszystkich struktur stawu – chrząstki stawowej, podchrzęstnych warstw kości, torebki stawowej, błony maziowej i struktur okołostawowych (ryc. 1 – str. 8).
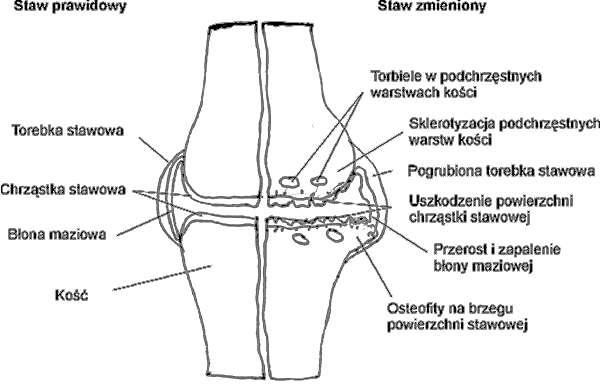
Ryc. 1. Zmiany stawowe w przebiegu choroby zwyrodnieniowej.
Chrząstka stawowa jest tkanką, która w niezwykły sposób łączy delikatność i prostotę struktury z wielką wytrzymałością. Chondrocyty są jedynym rodzajem komórek obecnych w chrząstce i stanowią 1-2% jej całkowitej objętości. Tworzą one w chrząstce skupiska, tzw. chondrony. Głównym, pod względem objętości, składnikiem chrząstki stawowej (98-99%) jest substancja międzykomórkowa (substancja podstawowa, matrix). Włókna kolagenu typu II (w mniejszym stopniu inne typy, m.in. IX i XI) tworzą rodzaj „rusztowania”, które nadaje chrząstce kształt, zapewnia spoistość i trwałość mechaniczną. Przestrzenie między włóknami wypełnione są wielkoczęsteczkowymi kompleksami białek i glikozaminoglikanów, przede wszystkim siarczanu chondroityny i siarczanu keratanu. Białka wiążące („link proteins”) łączą poszczególne kompleksy ze sobą i z cząsteczkami kwasu hialuronowego, tworząc rodzaj olbrzymich wielocząsteczkowych struktur. Chrząstka stawowa zawiera ponadto liczne dodatkowe składniki (tab. 1), które, mimo że występują w niewielkich ilościach, odgrywają istotną rolę w utrzymaniu jej struktury i funkcji. Drobnocząsteczkowe, bogate w leucynę proteoglikany (dekoryna, biglikan, fibromodulina, lumikan) wchodzą w interakcję z kolagenem i pomagają utrzymać jego strukturę. Ponadto mają one zdolność wiązania czynników wzrostowych, takich jak transforming growth factor-b (TGF-b) (2, 3). Dwa inne białka – białko bogate w prolinę, argininę i leucynę znane jako PRELP (proline/arginine-rich end leucine-rich repeat protein) i chondroadheryna uczestniczą w interakcji chondrocyt-substancja podstawowa chrząstki. Podobną rolę odgrywają ankoryna II, tenascyna i fibronektyna (4). Substancja podstawowa chrząstki decyduje o jej wytrzymałości na ściskanie i elastyczność, a także odgrywa rolę w utrzymaniu i regulacji zawartości płynu tkankowego.
Tabela 1. Białka substancji podstawowej chrząstki (3, 4).
| Białka kolagenowe | Proteoglikany | Białka niekolagenowe | Białka błonowe |
Typ II
Typ IX
Typ XI
Typ VI
Typ X (płytka wzrostowa)
Typ XIV | Agrekan
Biglikan
Dekoryna
Fibromodulina
Lumikan
Epifikan | COMP*
Matrylina 1
Ankoryna II
Tenascyna
Trombomodulina
PRELP*
Chondroadheryna
Fibronektyna | Syndekan
Perlekan
CD44
Integryny a (typ 1, 2, 3, 5, 6, 10)
Integryny b (typ 1, 3, 5) |
* COMP – oligomeryczne białko substancji podstawowej chrząstki (cartilage oligomeric matrix protein), PRELP – białko bogate w prolinę, argininę i leucynę (proline/arginine-rich end leucine-rich repeat protein).
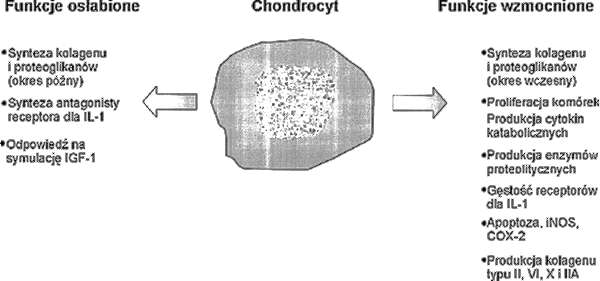
Ryc. 2. Zmiany fenotypu chondrocyta w przebiegu choroby zwyrodnieniowej. IL-1 – interleukina 1, IGF-1 – somatomedyna 1, iNOS – syntaza tlenku azotu, COX-2 – cyklooksygenaza-2.
Uszkodzenie chrząstki stawowej jest zjawiskiem pierwotnym w patogenezie choroby zwyrodnieniowej stawów. Lista czynników uszkadzających jest długa (tab. 2). Wszystkie one prowadzą do zaburzeń czynności chondrocytów, komórek o kluczowym znaczeniu dla integralności chrząstki. Zmiana fenotypu chondrocyta zaburza równowagę procesów anabolicznych i katabolicznych (ryc. 2), co z kolei jest przyczyną zmiany struktury, składu chemicznego i właściwości substancji podstawowej chrząstki.
Tabela 2. Klasyfikacja i przyczyny choroby zwyrodnieniowej stawów*.
| Pierwotna | Wtórna |
|
Z zajęciem stawów obwodowych
stawy międzypaliczkowe rąk
(postać guzkowa)
inne małe stawy (stawy śródręczno-paliczkowe, śródstopno-paliczkowy I)
stawy duże (staw kolanowy,
staw biodrowy)
Z zajęciem stawów kręgosłupa
stawy międzywyrostkowe
stawy międzykręgowe
Postaci szczególne
Postać zapalno-nadżerkowa
Postać uogólniona
Chondromalacja rzepki
Uogólniona hyperostoza szkieletowa (DISH)
| Związana z urazem
Postać ostra
Postać przewlekła (związana z zawodem,
aktywnością sportową, otyłością)
Związana z innymi chorobami stawów
Procesy miejscowe (złamania, martwica
jałowa, zakażenie)
Procesy uogólnione (reumatoidalne
zapalenie stawów, zespoły nadmiernej
wiotkości, osoczowe skazy krwotoczne)
Choroby metaboliczne
Alkaptonuria
Hemochromatoza
Choroba Wilsona
Choroba Kashin-Becka | Zaburzenia wydzielania wewnętrznego
Akromegalia
Nadczynność przytarczyc
Cukrzyca
Krystalopatie
Związane z kryształami dwuwodnego
pirofosforanu wapnia
Związane z kryształami hydroksyapatytu
wapnia
Neuropatie (stawy Charcot´a)
cukrzyca
wiąd rdzenia
Dysplazje kości
Inne zespoły |
* Zmodyfikowano wg Pelletier J-P, Martel-Pelletier J, Howell DS.: Etiopathogenesis of Osteoarthritis. In: William J. Koopman (Ed.): Arthritis and Allied Conditions. A Textbook of Rheumatology (14th edition), Lippincott Williams & Wilkins 2001.
Otyłość powoduje przeciążenie stawów kończyn dolnych i przyspiesza rozwój zmian zwyrodnieniowych stawów kolanowych (5). Osłabienie siły mięśniowej (6) i zaburzenia kontroli ruchowej (7) stanowią dodatkowe czynniki ryzyka. Przeciążenie i urazy związane z wykonywanym zawodem lub aktywnością sportową mogą być przyczyną uszkodzenia chrząstki stawowej i rozwoju zmian zwyrodnieniowych (8). Zmiany w chrząstce związane z procesem starzenia mogą odgrywać znaczną rolę w rozwoju zmian zwyrodnieniowych (9, 10, 11) i dotyczą wszystkich struktur stawu.
Pewne dane wskazują na rolę czynników dietetycznych. U chorych na chorobę zwyrodnieniową stawów kolanowych wykrywa się często obniżone stężenie aktywnych metabolitów witaminy D3, a spożywanie wysokich dawek antyoksydantów (kwas askorbinowy, b-karoten) może spowalniać postęp choroby (9). W badaniach in vitro a-tokoferol już w fizjologicznych stężeniach hamuje oksydację kolagenu i degradację substancji podstawowej chrząstki wskutek zahamowania utleniania lipidów w chondrocytach (13), a ograniczone dane kliniczne sugerują korzystny wpływ suplementacji witaminą E na przebieg choroby zwyrodnieniowej (14). W badaniach na modelach zwierzęcych niedobór witaminy B6 prowadzi do uszkodzenia chrząstki stawowej, wtórnego zapalenia stawów i zmniejszenia ilości dekoryny w zewnętrznych warstwach chrząstki stawowej (15). Wreszcie, czynniki dietetyczne odgrywają kluczową rolę w rozwoju szczególnej postaci choroby zwyrodnieniowej – choroby Kashin-Becka (16, 17).
Wrodzone anomalie układu ruchu o nieznanym podłożu (np. wrodzona dysplazja panewki stawu biodrowego, wrodzona szpotawość lub koślawość stawów) często prowadzą do rozwoju choroby zwyrodnieniowej (17). Wyniki najnowszych badań sugerują, że nieprawidłowe (koślawe lub szpotawe) ustawienie osi stawów kolanowych zwiększa 4- do 5-krotnie ryzyko progresji zmian zwyrodnieniowych odpowiednio po przyśrodkowej i bocznej stronie stawu, a szybkość progresji i narastania zaburzeń czynnościowych koreluje ze stopniem nasilenia wady (18). Zapalenia stawów, jak choćby najczęstsze z nich reumatoidalne zapalenie stawów, i zaburzenia wydzielania wewnętrznego (np. akromegalia) są znanymi przyczynami rozwoju zmian zwyrodnieniowych (20).
Choroba zwyrodnieniowa może rozwijać się w wyniku wrodzonych defektów struktury kolagenu, np. w zespole Ehlersa-Danlosa, a także w wyniku uwarunkowanych genetycznie defektów metabolicznych, prowadzących do gromadzenia się w tkankach nieprawidłowych metabolitów (np. hemochromatoza, ochronoza, choroba Gauchera, choroba Wilsona) (21).
Rola czynników genetycznych w patogenezie choroby zwyrodnieniowej nie ogranicza się do opisanych wyżej, dobrze poznanych anomalii. Badania bliźniąt jednojajowych wskazują, że czynniki genetyczne mogą determinować rozwój zmian zwyrodnieniowych stawów kolanowych i stawów rąk u kobiet w odpowiednio 39% i 65% (22). Silny wpływ dziedziczenia obserwuje się w przypadku choroby zwyrodnieniowej stawów biodrowych; występowanie zmian zwyrodnieniowych u jednego z bliźniąt jest związane z cztero- do sześciokrotnym wzrostem ryzyka wystąpienia zmian u drugiego z bliźniąt (23) i blisko dwukrotnym wzrostem ryzyka endoprotezoplastyki (24). Dotychczas nie zidentyfikowano genów odpowiedzialnych za dziedziczenie pierwotnej choroby zwyrodnieniowej. Co najmniej jeden z nich, odpowiedzialny za chorobę zwyrodnieniową stawu biodrowego, znajduje się na długim ramieniu chromosomu 2 (25). Gen lub geny odpowiedzialne za rozwój zmian zwyrodnieniowych mogą znajdować się także na długim ramieniu chromosomu 11 (26). Występowanie mutacji genu dla kolageu typu IX COL9A1 (6q12-q13) jest prawdopodobnie związane z podwyższonym ryzykiem rozwoju choroby zwyrodnieniowej stawów kolanowych i/lub biodrowych u kobiet (27). Znaczenie mutacji genów dla kolagenu typu I COL1A1, genu receptora witaminy D i genu receptora estrogenowego w rozwoju zmian zwyrodnieniowych nie jest jasne (28).
Działanie czynników uszkadzających prowadzi w pierwszej kolejności do uruchomienia mechanizmów naprawczych; we wczesnych okresach choroby zwyrodnieniowej obserwuje się proliferację chondrocytów ze wzrostem ich liczby w skupiskach i wzrost syntezy substancji podstawowej chrząstki (29). Wytwarzane glikozaminoglikany różnią się jednak pod względem budowy cząsteczki i właściwości od prawidłowych. W konsekwencji proces naprawczy nie zapewnia restytucji struktury i funkcji chrząstki stawowej, a nasilające się zaburzenia funkcji chondrocytów prowadzą do jej postępującej dezintegracji – chrząstka stawowa ulega ścieńczeniu, a jej powierzchnia staje się nierówna, pokryta drobnymi pęknięciami i szczelinami. W późnym okresie choroby coraz większa powierzchnia chrząstki ulega zniszczeniu, odsłaniając podchrzęstne warstwy kości.
Jeszcze do niedawna uważano, że obserwowane w przebiegu choroby zwyrodnieniowej pogrubienie i sklerotyzacja podchrzęstnych warstw kości są zjawiskiem wtórnym (30). Dziś przeważa pogląd, że zmiany w podchrzęstnych warstwach kości przebiegają równolegle, a czasem wyprzedzają zmiany w chrząstce; postępujące usztywnienie kości może sprzyjać powstawaniu nowych uszkodzeń chrząstki stawowej w wyniku nieprawidłowego rozkładu napięć mechanicznych lub mikrozłamań. Jak wykazano, nasilenie zjawiska sklerotyzacji podchrzęstnych warstw kości koreluje ze wzrostem jej sztywności (30). Postępowi sklerotyzacji nie towarzyszy natomiast adekwatny wzrost mineralizacji, co może wskazywać na zaburzenia procesu remodelizacji kości w chorobie zwyrodnieniowej stawów (31). Istotnie, szereg danych potwierdza istnienie zaburzeń czynności osteoblastów pochodzących z podchrzęstnych warstw kości chorych; wykryto m.in. nieprawidłową odpowiedź na stymulację parathormonem, witaminą D3, prostaglandyną E2 i niektórymi czynikami wzrostu, z konsekwencją w postaci zaburzeń produkcji białek kolagenowych i niekolagenowych, w tym osteokalcyny (32, 33, 34, 35).
Płyn stawowy, który dostaje się do podchrzęstnych warstw kości przez uszkodzenia jej powierzchni, powoduje osteolizę niektórych beleczek kości gąbczastej i przesunięcie innych w kierunku obwodowym, co prowadzi do powstania charakterystycznych dla choroby zwyrodnieniowej torbieli kostnych ze sklerotyczną otoczką (36).
Towarzyszący proces zapalny odgrywa istotną rolę w patomechanizmie choroby zwyrodnieniowej stawów. Cechy zapalenia błony maziowej w badaniu histopatologicznym wykrywa się u większości chorych (37, 38, 39). Obraz histopatologiczny błony maziowej chorych jest zwykle heterogenny; w obrębie jednego stawu obserwuje się cechy włóknienia tuż obok zmian zapalnych przypominających synovitis w przebiegu reumatoidalnego zapalenia stawów – hyperplazję komórek maziówki i nacieki zapalne złożone głównie z limfocytów T i monocytów. Sakkas i wsp. wykazali obecność CD3-pozytywnych limfocytów T w błonach maziowych 65% chorych na chorobę zwyrodnieniową oraz obecność komórek jednojądrzastych z ekspresją antygenów powierzchniowych wskazujących na stan aktywacji (CD 69, CD 25, CD 38, CD43, CD45RO i antygeny MHC II) (40). U około połowy chorych stwierdzono obecność transkryptów cytokin – interleukiny 2 (IL-2), interleukiny-10 (IL-10) i interferonu g (IFN-g) (rolę cytokin w patogenezie choroby zwyrodnieniowej opisano szczegółowo w dalszej części pracy). Zapalenie błony maziowej w chorobie zwyrodnieniowej ma charakter wtórny i jest przypuszczalnie związane z degradacją chrząstki stawowej i uwalnianiem jej fragmentów do płynu stawowego, które z kolei działają drażniąco na błonę maziową i indukują odpowiedź immunologiczną (41). Uwalniane przez komórki zapalne enzymy degradujące substancję podstawową chrząstki (patrz niżej) potęgują jej niszczenie, co prowadzi w efekcie do powstania mechanizmu „błędnego koła”.
Powstawanie osteofitów na brzegach powierzchni stawowych jest znamienną cechą choroby zwyrodnieniowej stawów. Przyczyna tego zjawiska nie została ostatecznie wyjaśniona; ograniczone dane wskazują na udział cytokin – interleukiny 1 (IL-1) i TGF-b (42), a także czynników genetycznych (43).
ROLA ENZYMÓW PROTEOLITYCZNYCH W DEGRADACJI SUBSTANCJI PODSTAWOWEJ CHRZĄSTKI STAWOWEJ
U chorych na chorobę zwyrodnieniową stawów powstaje szereg zmian w substancji podstawowej chrząstki – m.in. zmniejsza się zawartość siarczanu chondroityny, maleje masa cząsteczkowa i wzrasta rozpuszczalność glikozaminoglikanów chrząstki, wreszcie dochodzi do degradacji włókien kolagenowych (44). Wiodąca rola w powstawaniu opisanych zmian, obok czynników mechanicznych, przypada enzymom proteolitycznym. Najważniejsze z nich, tzw. metaloproteinazy (matrix metalloproteinases, MMPs), chociaż należą do tej samej grupy, różnią się pod względem swoistości i lokalizacji komórkowej.
Aktywność trzech kolagenaz (MMP-1, MMP-8, MMP-13) jest znacząco podwyższona w tkankach stawowych chorych na chorobę zwyrodnieniową (45, 46, 47, 48, 49, 50, 51). Chociaż wszystkie rozkładają włókna kolagenu typu II, różnią się właściwościami i lokalizacją tkankową. MMP-1 i MMP-8 występują głównie w powierzchownych warstwach chrząstki stawowej, podczas gdy MMP-13 wykrywana jest przede wszystkim w głębszych jej warstwach (49, 52, 53, 54). O ile aktywność dwóch pierwszych kolagenaz rośnie wraz z postępem choroby, aktywność MMP-13 utrzymuje się na stałym poziomie (53). MMP-1 występuje w dużych ilościach w błonie maziowej chorych (54). Uważa się, że MMP-1 i MMP-8 odgrywają rolę w procesach katabolicznych chrząstki stawowej, natomiast MMP-13 pełni funkcje związane z jej remodelizacją.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Dieppe P.: Osteoarthritis: time to shift the paradigm. This includes distinguishing between severe disease and common minor disability. BMJ 1999; 318(7194): 1299-1300. 2. Wiberg C. et al.: Biglycan and decorin bind close to the n-terminal region of the collagen VI triple helix. J. Biol. Chem. 2001; 276(22): 18947-18952. 3. Goldring M.B.: The role of the chondrocyte in osteoarthritis. Arthritis Rheum. 2000; 43(9): 1916-1926. 4. Knudson C.B., Knudson W.: Cartilage proteoglycans. Semin. Cell Dev. Biol. 2001; 12(2): 69-78. 5. Felson D.T.: Epidemiology of osteoarthritis. In: Brandt K.D., Doherty M., Lohmander L.S., eds. Osteoarthritis. Oxford: Oxford University Press; 1998:13-22. 6. Yang K.H. et al.: Diminished lower limb deceleration as a factor in early stage osteoarthritis. Trans. Orthop. Res. Soc. 1989; 14: 52 (abst). 7. Radin E.L. et al.: Relationship between lower limb dynamics and knee joint pain. J. Orthop. Res. 1991; 9(3): 398-405. 8. Buckwalter J.A.: Osteoarthritis and articular cartilage use, disuse, and abuse: experimental studies. J. Rheumatol. 1995; 43 (Suppl.): 13-15. 9. Huber M., et al.: Anatomy, biochemistry, and physiology of articular cartilage. Invest. Radiol. 2000; 35(10): 573-580. 10. Hamerman D.: Biology of the aging joint. Clin. Geriatr. Med. 1998; 14(3): 417-433.11. Kirkwood T.B.: What is the relationship between osteoarthritis and ageing? Baillieres Clin. Rheumatol. 1997; 11(4): 683-694. 12. McAlindon T., Felson D.T.: Nutrition: risk factors for osteoarthritis. Ann. Rheum. Dis. 1997; 56: 397-400. 13. Tiku M.L. et al.: Evidence linking chondrocyte lipid peroxidation to cartilage matrix protein degradation. Possible role in cartilage aging and the pathogenesis of osteoarthritis. J. Biol. Chem. 2000; 275(26): 20069-20076. 14. Sowers M., Lachance L.: Vitamins and arthritis. The roles of vitamins A, C, D, and E. Rheum. Dis. Clin. North. Am. 1999; 25(2): 315-332. 15. Masse P.G. et al.: Loss of decorin from the surface zone of articular cartilage in a chick model of osteoarthritis. Acta Histochem. 1997; 99(4): 431-444. 16. Sokoloff L.: The history of Kashin-Beck disease. NY State J. Med. 1989; 89: 343-351. 17. Utiger R.D.: Kashin-Beck disease – expanding the spectrum of iodine-deficiency disorders. N. Engl. J. Med. 1998; 339(16): 1156-1158. 18. Sharma L. et al.: The role of knee alignment in disease progression and functional decline in knee osteoarthritis. JAMA; 286(2): 188-195. 19. Murray R.O.: The aetiology of primary osteoarthritis of the hip. Br. J. Radiol. 1965; 38: 810-824. 20. Nuki G.: Osteoarthritis: a problem of joint failure. Z Rheumatol. 1999; 58(3): 142-147. 21. Balint G., Szebenyi B.: Hereditary disorders mimicking and/or causing premature osteoarthritis. Baillieres Best Pract. Res. Clin. Rheumatol. 2000; 14(2): 219-250. 22. Spector T.D. et al.: Genetic influences on osteoarthritis in women: a twin study. BMJ 1996; 312(7036): 940-943. 23. Lanyon P. et al.: Assessment of a genetic contribution to osteoarthritis of the hip: sibling study. BMJ 2000; 321(7270): 1179-1183. 24. Chitnavis J. et al.: Genetic influences in end-stage osteoarthritis. Sibling risks of hip and knee replacement for idiopathic osteoarthritis. J. Bone Joint Surg. Br. 1997; 79(4): 660-664. 25. Loughlin J. et al.: Linkage analysis of chromosome 2q in osteoarthritis. Rheumatology (Oxford) 2000; 39(4): 377-381. 26. Chapman K. et al.: Osteoarthritis-susceptibility locus on chromosome 11q, detected by linkage. Am. J. Hum. Genet. 1999; 65(1): 167-174. 27. Mustafa Z. et al.: Linkage analysis of candidate genes as susceptibility loci for osteoarthritis-suggestive linkage of COL9A1 to female hip osteoarthritis. Rheumatology (Oxford) 2000; 39(3): 299-306. 28. Loughlin J. et al.: Association analysis of the vitamin D receptor gene, the type I collagen gene COL1A1, and the estrogen receptor gene in idiopathic osteoarthritis. J. Rheumatol. 2000; 27(3): 779-784. 29. Adams M.E., Brandt K.D.: Hypertrophic repair of canine articular cartilage in osteoarthritis after anterior cruciate ligament transection. J. Rheumatol. 1991; 18(3): 428-435. 30. Li B., Aspden R.M.: Composition and mechanical properties of cancellous bone from the femoral head of patients with osteoporosis or osteoarthritis. J. Bone Miner. Res. 1997; 12(4): 614-651. 31. Grynpas M.D. et al.: Subchondral bone in osteoarthritis. Calcif Tissue Int 1991; 49(1): 20-26. 32. Hilal G. et al.: Osteoblast-like cells from human subchondral osteoarthritic bone demonstrate an altered phenotype in vitro: possible role in subchondral bone sclerosis. Arthritis Rheum. 1998; 41(5): 891-899. 33. Westacott C.I. et al.: Alteration of cartilage metabolism by cells from osteoarthritic bone. Arthritis Rheum. 1997; 40(7): 1282-1291. 34. Hilal G. et al.: Endogenous prostaglandin E2 and insulin-like growth factor 1 can modulate the levels of parathyroid hormone receptor in human osteoarthritic osteoblasts. J. Bone Miner. Res. 2001; 16(4): 713-721. 35. Hilal G. et al.: Abnormal regulation of urokinase plasminogen activator by insulin-like growth factor 1 in human osteoarthritic subchondral osteoblasts. Arthritis Rheum. 1999; 42(10): 2112-2122. 36. Resnick D. et al.: Subchondral cysts (geodes) in arthritic disorders: pathologic and radiographic appearance of the hip joint. AJR Am. J. Roentgenol. 1977; 128(5): 799-806. 37. Goldenberg D.L. et al.: Inflammatory synovitis in degenerative joint disease. J. Rheumatol. 1982; 9(2): 204-209. 38. Lindblad S., Hedfors E.: Arthroscopic and immunohistologic characterization of knee joint synovitis in osteoarthritis. Arthritis Rheum. 1987; 30 (10): 1081-1088. 39. Haraoui B. et al.: Synovial membrane histology and immunopathology in rheumatoid arthritis and osteoarthritis. In vivo effects of anti-rheumatic drugs. Arthritis Rheum. 1991; 34(2): 153-163. 40. Sakkas L.I. et al.: T cells and T-cell cytokine transcripts in the synovial membrane in patients with osteoarthritis. Clin. Diagn. Lab. Immunol. 1998; 5(4): 430-437. 41. Lohmander L.S.: The role of molecular markers to monitor breakdown and repair. In: Reginster J.Y., Pelletier J.P., Martel-Pelletier J., et al., eds. Osteoarthritis:. clinical and experimental aspects Berlin: Springer-Verlag; 1999: 296-311. 42. van Beuningen H.M. et al.: Transforming growth factor-beta 1 stimulates articular chondrocyte proteoglycan synthesis and induces osteophyte formation in the murine knee joint. Lab. Invest. 1994; 71(2): 279-290. 43. Uitterlinden A.G. et al.: Vitamin D receptor genotype is associated with radiographic osteoarthritis at the knee. J. Clin. Invest. 1997; 100(2): 259-263. 44. Hough A.J.: Pathology of osteoarthritis. In: Koopman W.J., ed. Arthritis and allied conditions,. 13th ed Philadelphia: Lea and Febiger; 1996: 1945-1968. 45. Martel-Pelletier J. et al.: Excess of metalloproteases over tissue inhibitor of metalloprotease may contribute to cartilage degradation in osteoarthritis and rheumatoid arthritis. Lab. Invest. 1994; 70(6): 807-815. 46. Reboul P. et al.: The new collagenase, collagenase-3, is expressed and synthesized by human chondrocytes but not by synoviocytes: a role in osteoarthritis. J. Clin. Invest. 1996; 97(9): 2011-2019. 47. Martel-Pelletier J., Pelletier J.P.: Wanted: the collagenase responsible for the destruction of the collagen network in human cartilage! Br. J. Rheumatol. 1996; 35(9): 818-820. 48. Mitchell P.G. et al.: Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase-13 from human osteoarthritic cartilage. J. Clin. Invest. 1996; 97(3): 761-768. 49. Chubinskaya S. et al.: Chondrocyte matrix metalloproteinase-8: up-regulation of neutrophil collagenase by interleukin-1 in human cartilage from knee and ankle joints. Lab. Invest. 1996; 74(1): 232-240. 50. Cole A.A. et al.: Chondrocyte matrix metalloproteinase-8. Human articular chondroctyes express neutrophil collagenase. J. Biol. Chem. 1996; 271(18): 11023-11026. 51. Shlopov B.V. et al.: Osteoarthritic lesions: involvement of three different collagenases. Arthritis Rheum. 1997; 40(11): 2065-2074. 52. Moldovan F. et al.: Collagenase-3 (matrix metalloprotease 13) is preferentially localized in the deep layer of human arthritic cartilage in situ: in vitro mimicking effect by transforming growth factor beta. Arthritis Rheum. 1997; 40(9): 1653-1661. 53. Fernandes J.C. et al.: Collagenase-1 and collagenase-3 synthesis in early experimental osteoarthritic canine cartilage. An immunohistochemical study. J. Rheumatol. 1998; 25(8): 1585-1594. 54. Nguyen Q. et al.: Preferential mRNA expression of prostromelysin relative to procollagenase and in situ localization in human articular cartilage. J. Clin. Invest. 1992; 89(4): 1189-1197. 55. Zafarullah M. et al.: Elevated metalloproteinase and tissue inhibitor of metalloproteinase mRNA in human osteoarthritic synovia. J. Rheumatol. 1993; 20(4): 693-697. 56. Walakovits L.A. et al.: Detection of stromelysin and collagenase in synovial fluid from patients with rheumatoid arthritis and posttraumatic knee injury. Arthritis Rheum. 1992; 35(1): 35-42. 57. Okada Y. et al.: Degradation of type IX collagen by matrix metalloproteinase 3 (stromelysin) from human rheumatoid synovial cells. FEBS Lett 1989; 244(2): 473-476. 58. Murphy G. et al.: Stromelysin is an activator of procollagenase. A study with natural and recombinant enzymes. Biochem. J. 1987; 248(1): 265-268. 59. Tetlow L.C. et al.: Matrix metalloproteinase and proinflammatory cytokine production by chondrocytes of human osteoarthritic cartilage: associations with degenerative changes. Arthritis Rheum. 2001; 44(3): 585-594. 60. Hembry R.M. et al.: Immunolocalisation studies on six matrix metalloproteinases and their inhibitors, TIMP-1 and TIMP-2, in synovia from patients with osteo- and rheumatoid arthritis. Ann. Rheum. Dis. 1995; 54(1): 25-32. 61. Yoshihara Y. et al.: Matrix metalloproteinases and tissue inhibitors of metalloproteinases in synovial fluids from patients with rheumatoid arthritis or osteoarthritis. Ann. Rheum. Dis.; 59(6): 455-461. 62. Ohuchi E. et al.: Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J. Biol. Chem. 1997; 272(4): 2446-2451. 63. Buttner F.H. et al.: Expression of membrane type 1 matrix metalloproteinase in human articular cartilage. Arthritis Rheum. 1997; 40(4): 704-709. 64. Imai K. et al.: Membrane-type matrix metalloproteinase 1 is a gelatinolytic enzyme and is secreted in a complex with tissue inhibitor of metalloproteinases 2. Cancer Res. 1996; 56(12): 2707-2710. 65. Caterson B. et al.: Mechanisms involved in cartilage proteoglycan catabolism. Matrix Biol. 2000; 19(4): 333-344. 66. Sandy J.D. et al.: Catabolism of aggrecan in cartilage explants. Identification of a major cleavage site within the interglobular domain. J. Biol. Chem. 1991; 266(14): 8683-8685. 67. Sandy J.D. et al.: The structure of aggrecan fragments in human synovial fluid. Evidence for the involvement in osteoarthritis of a novel proteinase which cleaves the Glu 373-Ala 374 bond of the interglobular domain. J. Clin. Invest. 1992; 89(5):1 512-1516. 68. Lohmander L.S. et al.: The structure of aggrecan fragments in human synovial fluid: evidence that aggrecanase mediates cartilage degradation in inflammatory joint disease, joint injury, and osteoarthritis. Arthritis Rheum. 1993; 36(9):1214-1222. 69. Lohmander L.S. et al.: Metalloproteinases, tissue inhibitor and proteoglycan fragments in knee synovial fluid in human osteoarthritis. Arthritis Rheum 1993; 36(2): 181-189. 70. Pendas A.M. et al.: Identification and characterization of a novel human matrix metalloproteinase with unique structural characteristics, chromosomal location, and tissue distribution. J. Biol. Chem. 1997; 272(7): 4281-4286. 71. Velasco G. et al.: Cloning and characterization of human MMP-23, a new matrix metalloproteinase predominantly expressed in reproductive tissues and lacking conserved domains in other family members. J. Biol. Chem. 1999; 274(8): 4570-4576. 72. Llano E. et al.: Identification and structural and functional characterization of human enamelysin (MMP-20). Biochemistry 1997; 36(49): 15101-15108. 73. Su S. et al.: Expression of the tissue inhibitors of metalloproteinases (TIMPs) gene family in normal and osteoarthritic joints. Rheumatol. Int. 1999; 18(5-6): 183-191. 74. Dean D.D. et al.: Evidence for metalloproteinase and metalloproteinase inhibitor imbalance in human osteoarthritic cartilage. J. Clin. Invest. 1989; 84(2): 678-685. 75. Nagase H Activation mechanisms of matrix metalloproteinases. Biol. Chem. 1997; 378(3-4): 151-160. 76. Quigley JP: Phorbol ester-induced morphological changes in transformed chick fibroblasts: evidence for direct catalytic involvement of plasminogen activator. Cell 1979; 17(1): 131-141. 77. Mochan E., Keler T.: Plasmin degradation of cartilage proteoglycan. Biochim. Biophys. Acta 1984; 800(2): 312-315. 78. Martel-Pelletier J. et al.: Plasmin, plasminogen activators and inhibitor in human osteoarthritic cartilage. J. Rheumatol. 1991; 18(12): 1863-1871. 79. Campbell I.K. et al.: Recombinant human interleukin-1 stimulates human articular cartilage to undergo resorption and human chondrocytes to produce both tissue- and urokinase-type plasminogen activator. Biochem. Biophys. Acta 1988; 967(2): 183-194. 80. Andrews J.L., Ghosh P.: Low molecular weight serine proteinase inhibitors of human articular cartilage. Isolation, characterization, and biosynthesis. Arthritis Rheum. 1990; 33(9): 1384-1393. 81. Eeckhout Y., Vaes G.: Further studies on the activation of procollagenase, the latent precursor of bone collagenase. Effects of lysosomal cathepsin B, plasmin and kallikrein, and spontaneous activation. Biochem. J. 1977; 166(1): 21-31. 82. Martel-Pelletier J. et al.: Cathepsin B and cysteine protease inhibitors in human OA: effect of intra-articular steroid injections. J. Orthop. Res. 1990; 8(3): 336-344. 83. Westacott C.I., Sharif M.: Cytokines in osteoarthritis: mediators or markers of joint destruction? Semin. Arthritis Rheum. 1996; 25(4): 254-272. 84. Goldring M.B.: The role of cytokines as inflammatory mediators in osteoarthritis: lessons from animal models. Connect Tissue Res. 1999; 40(1):1-11. 85. Webb G.R. et al.: Chondrocyte tumor necrosis factor receptors and focal loss of cartilage in osteoarthritis. Osteoarthritis Cartilage 1997; 5(6): 427-437. 86. Martel-Pelletier J. et al.: The interleukin-1 receptor in normal and osteoarthritic human articular chondrocytes. Identification as the type I receptor and analysis of binding kinetics and biologic function. Arthritis Rheum. 1992; 35(5): 530-540. 87. Jacques C. et al.: Posttranscriptional effect of insulin-like growth factor-I on interleukin-1beta-induced type II-secreted phospholipase A2 gene expression in rabbit articular chondrocytes. J. Clin. Invest. 1997; 99(8): 1864-1872. 88. Clancy R.M. et al.: The role of nitric oxide in inflammation and immunity. Arthritis Rheum. 1998; 41(7): 1141-1151. 89. Lotz M.: The role of nitric oxide in articular cartilage damage. Rheum. Dis. Clin. North Am. 1999; 25(2): 269-282. 90. Goldring M.B., Berenbaum F.: Human chondrocyte culture models for studying cyclooxygenase expression and prostaglandin regulation of collagen gene expression. Osteoarthritis Cartilage 1999; 7(4): 386-388. 91. Studer R. et al.: Nitric oxide in osteoarthritis. Osteoarthritis Cartilage 1999; 7(4): 377-379. 92. Raisz L.G.: Prostaglandins and bone: physiology and pathophysiology. Osteoarthritis Cartilage 1999; 7(4): 419-421. 93. Crofford L.J.: COX-2 in synovial tissues. Osteoarthritis Cartilage 1999; 7(4): 406-408. 94. Abramson S.B.: The role of COX-2 produced by cartilage in arthritis. Osteoarthritis Cartilage 1999; 7(4): 380-381. 95. Shalom-Barak T. et al.: Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-kappaB. J. Biol. Chem. 1998; 273(42): 27467-27473. 96. Olee T. et al.: IL-18 is produced by articular chondrocytes and induces proinflammatory and catabolic responses. J. Immunol. 1999; 162(2): 1096-1100. 97. Schlaak J.F. et al.: Different cytokine profiles in the synovial fluid of patients with osteoarthritis, rheumatoid arthritis and seronegative spondylarthropathies. Clin. Exp. Rheumatol. 1996; 14(2): 155-162. 98. Henrotin Y.E. et al.: Effects of exogenous IL-1 beta, TNF alpha, IL-6, IL-8 and LIF on cytokine production by human articular chondrocytes. Osteoarthritis Cartilage 1996; 4(3): 163-173. 99. Li W.Q., Zafarullah M.: Oncostatin M up-regulates tissue inhibitor of metalloproteinases-3 gene expression in articular chondrocytes via de novo transcription, protein synthesis, and tyrosine kinase- and mitogen-activated protein kinase-dependent mechanisms. J. Immunol. 1998; 161(9): 5000-5007. 100. Alaaeddine N. et al.: Inhibition of tumor necrosis factor alpha-induced prostaglandin E2 production by the antiinflammatory cytokines interleukin-4, interleukin-10, and interleukin-13 in osteoarthritic synovial fibroblasts: distinct targeting in the signaling pathways. Arthritis Rheum. 1999; 42(4): 710-718. 101. Joosten L.A. et al.: Role of interleukin-4 and interleukin-10 in murine collagen-induced arthritis. Protective effect of interleukin-4 and interleukin-10 treatment on cartilage destruction. Arthritis Rheum 1997; 40(2): 249-260. 102. van der Kraan P.M., van den Berg W.B.: Anabolic and destructive mediators in osteoarthritis. Curr. Opin. Clin. Nutr. Metab. Care. 2000; 3(3): 205-211. 103. van den Berg W.B.: The role of cytokines and growth factors in cartilage destruction in osteoarthritis and rheumatoid arthritis. Z Rheumatol. 1999; 58(3): 136-141. 104. Fernihough J.K. et al.: Local disruption of the insulin-like growth factor system in the arthritic joint. Arthritis Rheum. 1996; 39(9): 1556-1565. 105. van Beuningen H.M. et al.: Osteoarthritis-like changes in the murine knee joint resulting from intra-articular transforming growth factor-beta injections. Osteoarthritis Cartilage 2000; 8(1): 25-33. 106. Grodzicky T., Elkon K.B.: Apoptosis in rheumatic diseases. Am. J. Med. 2000; 108(1): 73-82. 107. Lotz M. et al.: Mechanisms of chondrocyte apoptosis. Osteoarthritis Cartilage 1999; 7(4): 389-391. 108. Aigner T. et al.: Apoptotic cell death is not a widespread phenomenon in normal aging and osteoarthritis human articular knee cartilage: a study of proliferation, programmed cell death (apoptosis), and viability of chondrocytes in normal and osteoarthritic human knee cartilage. Arthritis Rheum. 2001; 44(6): 1304-1312. 109. Gordon G.V. et al.: Autopsy study correlating degree of osteoarthritis, synovitis and evidence of articular calcification. J. Rheumatol. 1984; 11(5): 681-686. 110. Ali S.Y. et al.: Microcrystal deposition in cartilage and in osteoarthritis. Bone Miner. 1992; 17(2): 115-118. 111. Swan A. et al.: Submicroscopic crystals in osteoarthritic synovial fluids. Ann. Rheum. Dis. 1994; 53(7): 467-470. 112. Ryan L.M., Cheung H.S.: The role of crystals in osteoarthritis. In: Brandt K.D., ed. Osteoarthritis,. Rheumatic Diseases of America, vol 25 Philadelphia: W.B. Saunders; 1999: 257-267. 113. Karpouzas G.A., Terkeltaub R.A.: New developments in the pathogenesis of articular cartilage calcification. Curr. Rheumatol. Rep. 1999; 1(2): 121-7. 114. Inoue H. et al.: Production of neuropeptide substance P by synovial fibroblasts from patients with rheumatoid arthritis and osteoarthritis. Neurosci Lett.; 303(3): 149-152. 115. Saito T., Koshino T.: Distribution of neuropeptides in synovium of the knee with osteoarthritis. Clin. Orthop. 2000; (376):172-182. 116. Menkes C.J. et al.: Substance P levels in the synovium and synovial fluid from patients with rheumatoid arthritis and osteoarthritis. J. Rheumatol. 1993; 20(4): 714-717. 117. Koff W.C., Dunegan M.A.: Modulation of macrophage-mediated tumoricidal activity by neuropeptides and neurohormones. J. Immunol. 1985; 135(1): 350-354. 118. Lotz M. et al.: Substance P activation of rheumatoid synoviocytes: neural pathway in pathogenesis of arthritis. Science 1987; 235(4791): 893-895. 119. Vilensky J.A., Cook J.A.: Neurogenic acceleration of osteoarthritis. Curr. Opin. Rheumatol. 1998; 10(3): 251-255. 120. Laurenzi M.A. et al.: Stimulation of human B lymphocyte differentiation by the neuropeptides substance P and neurokinin A. Scand. J. Immunol. 1989; 30(6): 695-701. 121. Matsuda H. et al.: Substance P induces granulocyte infiltration through degranulation of mast cells. J. Immunol. 1989; 142(3): 927-931. 122. Kimball E.S. et al.: Substance P, neurokinin A, and neurokinin B induce generation of IL-1-like activity in P388D1 cells. Possible relevance to arthritic disease. J. Immunol. 1988; 141(10): 3564-3569. 123. Kimball E.S., Fisher M.C.: Potentiation of IL-1-induced BALB/3T3 fibroblast proliferation by neuropeptides. J. Immunol. 1988; 141(12): 4203-4208.124. Arnalich F. et al.: Neuropeptides and interleukin-6 in human joint inflammation relationship between intraarticular substance P and interleukin-6 concentrations. Neurosci. Lett.; 170(2): 251-254. 125. Raap T. et al.: Neurotransmitter modulation of interleukin 6 (IL-6) and IL-8 secretion of synovial fibroblasts in patients with rheumatoid arthritis compared to osteoarthritis. J. Rheumatol.; 27(11): 2558-2565. 126. Chikanza I., Fernandes L.: Novel strategies for the treatment of osteoarthritis. Expert Opin. Investig. Drugs. 2000; 9(7): 1499-1510.

