Janusz Urban
Wybrane zagadnienia genodermatoz wieku rozwojowego – rybia łuska wrodzona i dziedziczna*
Selected problems of developmental age genodermatoses – ichthyosis congenital and hereditary
z Katedry i Kliniki Dermatologii, Wenerologii i Dermatologii Dziecięcej Akademii Medycznej w Lublinie
Kierownik Katedry: prof. dr hab. n. med. Barbara Lecewicz-Toruń
Streszczenie
Summary
The author discusses the aetiology and pathogenesis, clinical manifestations, diagnosis and complications related to generalized epidermal dyskeratosis.

Genodermatozy związane z uogólnionym zaburzonym rogowaceniem charakteryzują się nadmiernym rogowaceniem i złuszczaniem naskórka. W grupie tej wyróżniane są zazwyczaj: rybia łuska wrodzona (ichthyosis congenita), rybia łuska dziedziczna (ichthyosis hereditaria) oraz inne odmiany zaburzeń rogowacenia określane zwyczajowo rybią łuską (ichthyosis) np. rybia łuska linijna okrążająca Comela (ichthyosis linearis circumflexa Comel) (22, 38, 41) lub erytrokeratodermią np. erytrokeratodermia zmiennopostaciowa Gottrona (erythrokeratodermia figurata variabilis) oraz inne (41, 42). Osobną dużą grupę chorób związanych z zaburzonym rogowaceniem stanowią liczne odmiany nadmiernego dziedzicznego rogowacenia dłoni i podeszew (41, 43, 44).
Wspólną cechą rybiej łuski, zarówno wrodzonej (ichthyosis congenita) jak i rybiej łuski dziedzicznej (ichthyosis hereditaria), jest występowanie uogólnionego rozlanego zaburzonego rogowacenia naskórka o cechach nadmiernego rogowacenia – orthohyperkeratosis, rzadko niepełnego rogowacenia – parakeratosis. Choroby te, ze względu na pewne podobieństwo do wyglądu łusek ryby, określone zostały nazwą rybiej łuski (ichthyosis). Określenie to jest stosowane zwyczajowo dla opisania tej choroby. Nie obrazuje ono w pełni wyglądu zmienionej skóry i naskórka chorego, gdyż łuski naskórka nie nakładają się na siebie lecz ułożone są obok siebie porozdzielane rozpadlinami. Swoim wyglądem przypominają bardziej skórę gada, niż łuski ryby (30). Ten obraz szczególnie widoczny jest w rybiej łusce wężowatej (ichthyosis serpentina), która jest odmianą ciężką rybiej łuski czerniejącej, s. rybiej łuski X – chromosomalnej (ichthyosis nigricans, s. X-linked ichthyosis), rybiej łusce arlekinowej (ichthyosis harlequin), rybiej łusce blaszkowatej klasycznej (ichthyosis lamallaris classica) oraz rybiej łusce jeżastej (ichthyosis hystrix).
Zarówno rybia łuska wrodzona jak i dziedziczona powstają w następstwie genetycznych uszkodzeń lub mutacji genów oraz powstających w związku z tym zaburzeń wielu organelli komórkowych naskórka, skóry właściwej i innych tkanek. Warunkują one defekty strukturalne naskórka oraz zaburzenia biochemiczne lipidów warstwy rogowej i keratyn naskórkowych (6, 7, 10, 12, 13, 14, 15, 23, 29, 31, 33, 34, 35). Zaburzenia rogowacenia naskórka w rybiej łusce są rozlane i dotyczą całej skóry, zarówno skóry gładkiej – twarzy, szyi, tułowia i kończyn, owłosionej skóry głowy, dłoni, podeszew oraz przydatków skóry – płytek paznokciowych i włosów – zespół Nethertona oraz inne (22, 38, 39). Obraz kliniczny rybiej łuski jest różny w zależności od odmiany, poza tym wykazuje różnice fenotypowe. We wszystkich odmianach rybiej łuski wrodzonej (ichthyosis congenita) i rybiej łuski dziedzicznej (ichthyosis hereditaria), poza rybią łuską czerniejącą s. rybią łuską X – chromosomalną, zmiany na dłoniach i podeszwach występują jako wyraz uogólnionego nadmiernego rogowacenia naskórka z różnym fenotypowym nasileniem i pojawiają się w tym samym czasie co zmiany w innych okolicach skóry. Cecha ta odróżnia rybią łuskę wrodzoną i rybią łuskę pospolitą od idiopatycznego dziedzicznego nadmiernego rogowacenia skóry dłoni i podeszew, w których zmiany na dłoniach i podeszwach występują pierwotnie. Mogą one ograniczać się do wyłącznego występowania w tych miejscach lub też występują cechy transgrediencji tj. występowania ognisk rogowacenia na grzbietach rąk i stóp, okolicach łokci, kolan oraz innych miejscach twarzy, tułowia i kończyn, które pojawiają się zazwyczaj później (40, 41, 43, 44).
Odmiany rybiej łuski wrodzonej i dziedzicznej w niniejszym opracowaniu przedstawiono zgodnie z ogólnie przyjętą klasyfikacją w oparciu o następujące cechy chorobowe: 1) sposób dziedziczenia, 2) okres wystąpienia zmian skórnych, 3) obraz kliniczny zmian chorobowych, 4) obraz zmian histopatologicznych, histochemicznych i ultrastrukturalnych oraz 5) występowanie zaburzeń enzymatycznych i biochemicznych.
1. Rybia łuska wrodzona (ichthyosis congenita)
1.1. Rybia łuska arlekinowa (ichthyosis harlequin)
Rybia łuska arlekinowa (ichthyosis harlequin) jest ciężką postacią rybiej łuski wrodzonej (ichthyosis congenita gravis) (6). Jest ona synonimem używanego uprzednio określenia płód arlekinowy (harlequin fetus). Choroba ta charakteryzuje się nadmiernym rogowaceniem naskórka, rozpoczynającym się w życiu płodowym. Rybia łuska arlekinowa w ubiegłych latach była opisywana pod różnymi nazwami – ichthyosis congenita fetalis, ichthyosis intrauterina, keratosis diffusa fetalis, keratoma malignum congenitum i aligator baby (28).
Etiopatogeneza. Choroba ta jest genodermatozą, która występuje rodzinnie (28, 37) i dziedziczy się autosomalnie recesywnie, podobnie jak zespół dziecka kolodionowego (syndroma collodion baby). Odróżnia się od niej wybitnym nadmiernym rogowaceniem naskórka i ciężkim stanem ogólnym noworodka.
Przyczyny zaburzeń nadmiernego rogowacenia dotychczas nie zostały dokładnie poznane. Ultrastrukturalne badania wskazują, iż ichthyosis harlequin jest genetycznie różnorodną grupą zaburzeń ze zmienionymi blaszkowatymi ziarnami i międzykomórkowymi lipidami oraz zmienioną ekspresją strukturalnych proteinowych markerów naskórkowej kerytynizacji. Wykazano defekt w tworzeniu ciał blaszkowatych oraz powiększenie komórek warstwy rogowej z nagromadzeniem licznych struktur pęcherzykowych, które nie były wydzielane do warstwy rogowej. Podobne zmiany ultrastrukturalne stwierdzane są we wrodzonej erytrodermii rybiołuskowatej suchej (erythrodermia ichthyosiformis congenita sicca). Brak ciał blaszkowatych prowadzi do zaburzeń w degradacji komórek warstwy rogowej i zachowania tendencji hiperrozrostowej – nadmiernego rogowacenia (6, 12, 23).
Objawy kliniczne. Zmiany chorobowe występują przy urodzeniu. Rybia łuska arlekinowa występuje znacznie rzadziej w porównaniu z postacią łagodną rybiej łuski wrodzonej jaką jest zespół dziecka kolodionowego (syndroma collodion baby) (9, 28). Noworodki rodzą się niekiedy martwe, często przedwcześnie z niską masą urodzeniową ciała. Cała skóra pokryta jest twardą, bardzo grubą warstwą nadmiernie zrogowaciałego naskórka, barwy szarożółtej lub żółtowoskowej, która swoim wyglądem przypomina pancerz lub skórę krokodyla. Liczne pęknięcia warstwy rogowej powstają przy ruchach noworodka. Tworzą się różnej wielkości blaszki lub grube płytki rogowe kształtu prostokątnego, romboidalnego i wielobocznego porozdzielane rozpadlinami sięgającymi niekiedy do skóry właściwej. Wygląd zmienionego naskórka przypomina strój arlekina, z którym związana jest dawna nazwa płód arlekinowy (harlequin fetus) lub podobna jest do skóry krokodyla (aligator baby). Powstałe rozpadliny stanowią wrota dla wtórnych zakażeń bakteryjnych Staphylococcus aureus i Psudomonas aeruginosa, które mogą być przyczyną uogólnionego zakażenia. W następstwie nadmiernego rogowacenia naskórka twarz noworodka wykazuje zniekształcające zmiany pseudodysmorficzne. Nos może być spłaszczony, małżowiny uszne niekiedy zniekształcone lub niedorozwinięte z czopami rogowymi zamykającymi naturalne otwory tych narządów. Rogowacenie skóry policzków, nosa, czoła oraz powiek powoduje wywinięcie powiek (ectropion) i warg (eclabium). Ten stan utrudnia ssanie. Dzieci te wymagają karmienia przez zgłębnik lub drogą dożylną. Kończyny znajdują się w przykurczu z powodu nawarstwień rogowych nad stawami. Nawarstwienia rogowe powodują usztywnienie i utrudnienie ruchów w stawach palców rąk, nadgarstków oraz stóp. Palce u tych noworodków mogą wykazywać również niedorozwój i zniekształcenia, a niekiedy dochodzić może do samoistnej ich amputacji (ainhum). Nadmierne rogowacenie na tułowiu i brzuchu, tworzące rogowy pancerz utrudnia ruchomość klatki piersiowej, a tym samym oddychanie. Stan ogólny noworodków jest ciężki ze względu na zaburzenia czynnościowe wieloukładowe oraz dołączające się wtórne infekcje bakteryjne (6, 28). W dalszym przebiegu choroby płytki rogowe ulegają całkowitemu złuszczeniu i ukazuje się wówczas żywoczerwona erytrodermiczna skóra. Rogowacenie naskórka utrzymuje się nadal, jednak jego intensywność jest zmniejszona a wytwarzane łuski są drobniejsze i cieńsze. Obraz chorobowy w tym okresie ma cechy wrodzonej erytrodermii rybiołuskowatej suchej (erythrodermia ichthyosiformis congenita sicca) z podobnym obrazem zmian chociaż fenotypowo bardziej nasilonymi, jakie obserwowane są po ustąpieniu zmian u noworodków z syndroma collodion baby.
Rozpoznanie choroby ustalamy w oparciu o obraz kliniczny, występowanie niekiedy analogicznych zmian skórnych u rodzeństwa oraz badanie histopatologiczne ze zgrubieniem warstwy rogowej o cechach zbitej orthohyperkeratosis oraz z rogowaceniem ujść mieszków włosowych z towarzyszącymi cechami granulosis, acanthosis i papillomatosis oraz naciekami jednojądrzastymi wokół rozszerzonych naczyń powierzchownych skóry właściwej.
1.2. Zespół dziecka kolodionowego (Syndroma collodion baby)
Określenie dziecko kolodionowe (collodion baby) jest pojęciem używanym do opisania stanu noworodka, którego skóra po urodzeniu pokryta jest zgrubiałym naskórkiem o wyglądzie błony kolodionowej. Larregue i wsp. (18, 19) wykazali dużą zmienność fenotypową obrazu klinicznego zmian i dalszego przebiegu choroby u tych noworodków, stąd też uznali za właściwe zastosowanie określenia – zespół dziecka kolodionowego (syndroma collodion baby). Obserwowane przy urodzeniu zmiany są również następstwem uogólnionego nadmiernego rogowacenia naskórka w okresie życia płodowego, jednak mniejszego stopnia niż występujące w rybiej łusce arlekinowej. Zespół tych zmian chorobowych w większości przypadków jest początkowym okresem przewlekłych stanów chorobowych obserwowanych w latach późniejszych, o cechach wrodzonej erytrodermii rybiołuskowatej suchej (erythrodermia ichthyosiformis congenita sicca) lub rybiej łuski blaszkowatej (ichthyosis lamellaris) (10, 11, 17, 18, 19, 20, 24, 45). Liczni autorzy obserwowali również całkowitą regresję błony kolodionowej do stanu prawidłowej skóry bez objawów zaburzonego rogowacenia (8, 18, 19). Larregue i wsp. (18, 19) w grupie 267 noworodków z zespołem dziecka kolodionowego stwierdzali również występowanie innych chorób, a mianowicie: rybiej łuski linijnej okrążającej (ichthyosis linearis circumflexa Comel), zespołu Sjögrena-Larssona (syndroma Sjögren-Larsson), trichothiodystrophii, oraz choroby Conradiego (morbus Conradi, s. morbus Conradi-Hünermann).
Syndroma collodion baby jest uważana za łagodną postać rybiej łuski wrodzonej (ichthyosis congenita mitis), w odróżnieniu od postaci ciężkiej (ichthyosis congenita gravis), jaką jest rybia łuska arlekinowa (ichthyosis harlequin).
Etiopatogeneza.Syndroma collodion baby jest genodermatozą, którą dziedziczy się w sposób autosomalny recesywny (8). Częstość występowania określana jest 1:50 000 urodzeń. Zespół ten stwierdzany jest u 1/4 (20) lub u większości (27) noworodków przedwcześnie urodzonych z masą ciała poniżej 3000 g, bez predyspozycji płci w występowaniu choroby. Zmiany chorobowe stwierdzane były u rodzeństwa (braci lub sióstr) w 51%; natomiast nie stwierdzano ich u żadnego z rodziców chorych dzieci (20).
Objawy kliniczne. Noworodek przy urodzeniu wykazuje objawy uogólnionego nadmiernego rogowacenia naskórka. Skóra całego ciała pokryta jest zgrubiałym naskórkiem ściśle przylegającym do skóry, o wyglądzie przezroczystej błony przypominającej celofan lub wyschnięty kolodion. Zmianom tym towarzyszy widoczny przez napięty, przezroczysty naskórek, intensywny rumień skóry, który jest również charakterystyczną cechą tego zespołu. Naskórek „kolodionowy” ma wzmożoną konsystencję, jest sztywny w dotyku i robi wrażenie laminowanego papieru. Prześwitujący przez naskórek rumień skórny może być słabo widoczny w miejscach zwiększonego rogowacenia w związku ze zgrubieniem naskórka, który w tych miejscach ma wygląd grubego nieprzejrzystego pergaminu, barwy szarej lub szarożółtej. W obrębie skóry twarzy nasilone rogowacenie powoduje utrudnienie mimiki oraz powoduje zniekształcenie rysów twarzy. Widoczne jest wywinięcie powiek (ectropion) z obrzękiem spojówek powiekowych z poszerzeniem i niedomykalnością szpar powiekowych, oraz wywinięciem warg (eclabium) z rozwarciem ust w kształcie litery O, przypominające „pyszczek rybi”. W niektórych przypadkach nasilone rogowacenie obejmuje małżowiny uszne i powoduje ich niedorozwój lub zniekształcenie (ryc. 1), oraz deformację kształtu nosa ze zwężeniem zewnętrznych ujść przewodów tych narządów. Ruchomość kończyn górnych i dolnych jest ograniczona, niekiedy znajdują się w przykurczu. Ograniczona jest również ruchomość palców rąk. Paznokcie mają wygląd prawidłowy, włosy są niezmienione i przechodzą przez zgrubiały naskórek kolodionowy (18, 19, 27, 41, 45).

Ryc. 1. Zespół dziecka kolodionowego (Syndroma collodion baby). Widoczna błona kolodionowa z licznymi rozpadlinami na tułowiu oraz nawarstwieniami rogowymi na twarzy powodującymi występowanie ectropion i eclabium.
Przebieg choroby. W następstwie wykonywanych przez dziecko ruchów, dochodzi do pękania błony kolodionowej z powstawaniem powierzchownych rozpadlin, które w okolicach pachowych, pachwinowych oraz na szyi i nad stawami mogą sięgać skóry właściwej. Błona kolodionowa w następstwie pojawiających się pęknięć oraz stosowanych leków nawilżających i natłuszczających złuszcza się dużymi płatami. Złuszczanie może trwać od kilku dni do kilku tygodni w zależności od stopnia nasilenia objawów nadmiernego rogowacenia stwierdzanego przy urodzeniu (18, 19).
Powikłania. W pierwszych dobach życia noworodka dochodzić może do groźnych powikłań, do których należą przede wszystkim zaburzenia w utrzymaniu stałej ciepłoty ciała (skłonność do hipotermii) oraz zaburzenia wodno-elektrolitowe z odwodnieniem i hipernatremią (3). Do częstych powikłań należy również zachłystowe zapalenie płuc. Liczne rozpadliny naskórka i skóry stanowią wrota dla wtórnych zakażeń bakteryjnych, przede wszystkim Staphylococcus aureus i Pseudomonas aeruginosa oraz innych Gram-ujemnych bakterii, które mogą prowadzić do uogólnienia procesu zakaźnego z posocznicą włącznie. Spotykane są również zakażenia drożdżakami i grzybami. Zachodzi wówczas konieczność zastosowania leczenia przeciwbakteryjnego lub przeciwgrzybiczego miejscowego lub ogólnego, z uwzględnieniem wrażliwości wyhodowanych patogenów.
Rozpoznanie syndroma collodion baby jest łatwe, ze względu na charakterystyczny obraz kliniczny. Potwierdzeniem rozpoznania jest również obraz histopatologiczny wykazujący zgrubienie wszystkich warstw naskórka z cechami orthohyperkeratosis, granulosis acanthosis i papillomatosis. Rogowacenie widoczne jest również w ujściach mieszków włosowych.
Rozpoznanie różnicowe. Zespół dziecka kolodionowego (syndroma collodion baby) wymaga zróżnicowania z postacią ciężką rybiej łuski wrodzonej (ichthyosis harlequin, s. harlequin fetus), która wykazuje większe objawy rogowacenia oraz ciężki stan ogólny.
1.3. Wrodzona erytrodermia rybiołuskowata sucha (Erythrodermia ichthyosiformis congenita sicca)
Wrodzona erytrodermia rybiołuskowata sucha (erythrodermia ichthyosiformis congenita sicca – EICS) jest chorobą, która charakteryzuje się uogólnionym nadmiernym rogowaceniem naskórka z erytrodermią (ryc. 2). Zmiany obejmują całą skórę z zajmowaniem okolic pachowych, pachwinowych oraz fałdów skóry, zgięć i dołów nad stawami oraz dłonie i podeszwy stóp.
Etiopatogeneza. Choroba dziedziczy się w sposób autosomalny recesywny. Objawami poprzedzającymi obraz EICS jest najczęściej syndroma collodion baby (SCB). Rzadziej choroba ta rozpoczyna się stanem erytrodermicznym bez wyprzedzających objawów SCB. Przynależność klasyfikacyjną tej postaci rybiej łuski do EICS poza charakterystycznym obrazem klinicznym potwierdza wzrost wartości N-alkanów w naskórku (10, 47). Haftek i wsp. (10) w oparciu o badania wykonane w 1993 r. uważają, iż charakterystyczne zwiększenie poziomu N-alkanów w EICS jest wewnątrzpochodne i nie jest następstwem miejscowego stosowania leków. W tej chorobie stwierdzane jest również znaczne zwiększenie kinetyki komórek naskórka w porównaniu z rybią łuską blaszkowatą klasyczną (ichthyosis lamellaris classica – ILC). W badaniach wykonanych przez Hazella i Marksa (13), średni wskaźnik autoradiograficznego znakowania trytowaną tymidyną wykazywał znacznie wyższy wzrost dla tej choroby (EICS) i wynosił 19,70 ± 5,84, natomiast u chorych z ILC był niższy i wynosił 9,36 ± 4,05.
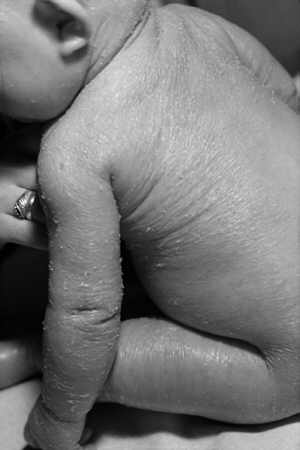
Ryc. 2. Wrodzona erytrodermia rybiołuskowata sucha (Erythrodermia ichthyosiformis congenita sicca). Dziewczynka w wieku 6 mies. urodzona z cechami syndroma collodion baby. Widoczny uogólniony rumień skóry z drobnymi jasnymi i biało-szarymi łuskami.
Objawy kliniczne. Choroba wykazuje charakterystyczne cechy, które odróżniają ją od rybiej łuski blaszkowatej (ILC). Hazell i Marks (13) oraz Wiliams i Elias (47) uważają, iż dla EICS wyróżniającymi objawami są:
1. Występowanie erytrodermii o różnej intensywności, która jest najbardziej charakterystyczną cechą tej choroby.
2. Obecność uogólnionego nadmiernego rogowacenia naskórka o łagodnym przebiegu z jasnymi lub jasnoszarymi łuskami z zajęciem również okolic pachowych, pachwinowych, dołów łokciowych, oraz innych fałdów skóry. Dłonie i podeszwy wykazują również rozlane rogowacenie naskórka o łagodnym nasileniu. Na podudziach mogą występować niekiedy duże, ciemne płytkowe łuski.
3. Wywinięcie powiek (ectropion) jest częste. W przypadkach z łagodnymi zmianami erytrodermicznymi skóra twarzy wykazywać może tylko zwiększone napięcie i suchość bez wywinięcia powiek.
4. Przebieg choroby wykazywać może zmienne nasilenie, niekiedy z poprawą w okresie dojrzewania.
5. Badanie histopatologiczne. Warstwa rogowa naskórka jest zgrubiała o cechach orthohyperkeratosis i składa się z cienkich rogowych blaszek ściśle przylegających do siebie. Warstwa komórek ziarnistych najczęściej o niezmienionej grubości. Miejscami występują ogniska parakeratosis z agranulosis. Warstwa komórek kolczystych z wyraźnymi cechami acanthosis. W skórze właściwej wokół brodawkowych i podbrodawkowych naczyń krwionośnych stwierdzane są nacieki jednojądrzaste (13, 47).
1.4. Rybia łuska blaszkowata klasyczna (Ichthyosis lamellaris classica)
Rybia łuska blaszkowata klasyczna (ichthyosis lamellaris classica – ILC) jest chorobą, która charakteryzuje się uogólnionym nadmiernym rogowaceniem naskórka. Dziedziczy się autosomalnie recesywnie. Chorobę poprzedzają objawy syndroma collodion baby lub ichthyosis harlequin.
Etiopatogeneza. Choroba ta dziedziczy się autosomalnie recesywnie. Opisano również przypadki z dziedziczeniem autosomalnym dominującym, w których skórne objawy chorobowe ujawniały się po pewnym okresie czasu od urodzenia. Zaburzenia molekularne w tej chorobie dotyczą genu transglutaminazy-1 (TG-1), który zlokalizowany jest w chromosomie 14q11 (15, 33). Brak aktywności TG-1, wykazany w badaniach naskórka zmienionego chorobowo warunkuje zaburzenie rogowacenia z nieprawidłowym tworzeniem tak zwanej zrogowaciałej koperty (cornified envelope). Zaburzona struktura rogowaciejącej komórki naskórka warunkuje nieprawidłowe oddzielanie się warstwy komórek rogowych. Poza tym u tych chorych w naskórku obserwowane jest obniżenie ekspresji involukryny o nieprawidłowym dyfuzyjnym ułożeniu w cytoplazmie komórek warstwy ziarnistej (36).
Objawy kliniczne. Omawiana choroba w odróżnieniu od EICS wykazuje inny obraz kliniczny oraz przewlekły postępujący przebieg z nasilaniem się zmian chorobowych z wiekiem. U tych chorych obserwuje się przeważające objawy nadmiernego ortohiperkeratotycznego rogowacenia z łagodnym rumieniem skóry, który nie zawsze jest widoczny. Główną cechą jest nadmierne rogowacenie z dużymi, grubymi łuskami barwy od ciemnoszarej do ciemnobrązowej, kształtu prostokątnego, wielobocznego lub romboidalnego. Szczególnie nasilone rogowacenie występuje w fałdach skóry, dołach pachowych, łokciowych i podkolanowych (ryc. 3a, b). W tych okolicach występuje zliszajowacenie skóry. Nasilone objawy rozlanego nadmiernego rogowacenia widoczne są również na rękach i stopach zarówno na powierzchniach grzbietowych jak i na dłoniach i stopach. Skóra twarzy wykazuje rogowacenie dużego stopnia, intensywniejsze niż w EICS, z częstym wywinięciem powiek (ectropion). Stałą cechą jest nadmierna suchość skóry. Płytki paznokciowe rąk i stóp u tych chorych wykazują zgrubienie (13, 36, 47).
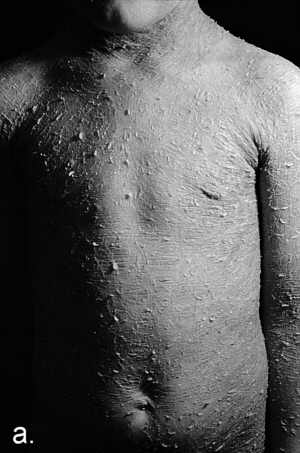
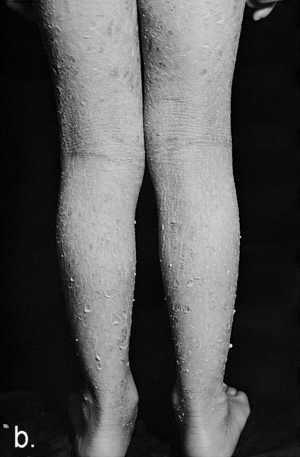
Ryc. 3a. Rybia łuska blaszkowata klasyczna (Ichthyosis lamellaris classica). Chłopiec w wieku 8 lat z wybitnie nasilonymi objawami uogólnionego nadmiernego rogowacenia z widocznymi dużymi ciemnymi łuskami na tułowiu (a) oraz w dołach podkolanowych (b).
Badanie histopatologiczne. Bardzo charakterystyczną histologiczną cechą dla ILC jest wybitny przerost warstwy rogowej naskórka o cechach orthohyperkeratosis, którego grubość bywa dwu- lub trzykrotnie większa niż u chorych z EICS. Zmianom tym towarzyszy również rogowacenie ujść mieszków włosowych. Warstwa komórek ziarnistych wykazuje wyraźne zgrubienie (granulosis) z kilkukrotnym zwielokrotnieniem warstw tych komórek. Ogniska parakeratozy u tych chorych stwierdzane są sporadycznie. Warstwa Malpighiego bez wyraźnych cech akantozy lub z niewielkim zgrubieniem warstwy komórek kolczystych (13, 36, 47).
Rozpoznanie omawianych chorób: EICS i ILC uważanych obecnie za odrębne jednostki chorobowe opiera się na odmiennych cechach klinicznych, charakterystycznych dla tych chorób, innym obrazie histopatologicznym, zmienionej kinetyce komórek naskórka oraz badaniach biochemicznych N-alkanów w naskórku.
Rozpoznanie różnicowe. Zarówno EICS jak i ILC wymagają zróżnicowania z następującymi postaciami rybiej łuski wrodzonej i dziedzicznej:
1. Nadmierne rogowacenie epidermolityczne, s. wrodzona erytrodermia rybiołuskowata pęcherzowa Brocqa (hyperkeratosis epidermolytica, s. erythrodermia ichthyosiformis congenita bullosa Brocq).
2. Rybia łuska czerniejąca, s. rybia łuska X – chromosomalna (ichthyosis nigicans, s. X – linked ichthyosis).
3. Rybia łuska pospolita (ichthyosis vulgaris).
4. Zespół złuszczającej się skóry (skin peeling syndrome).
5. Zespół Nethertona (syndroma Netherton).
6. Rogowacenie ciemne łagodne (acanthosis nigricans benigna).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Braun-Falco O. et al. (red.): Dermatology. Springer-Verlag, Berlin Heidelberg 1991, 511-529. 2. Brusasco A. et al.: Congenital reticular ichthyosiform erythroderma: a distinct type of ichthyosis. Proceedings of the 7th International Congress of Pediatric Dermatology, Buenos Aires, September 27 – October 1, 1994. Pierini A.-M. Garcia-Diaz de Pierini R., Bustamante R.E. (red.), Elsevir, Amsterdam, Lausanne, New York, Oxford, Shannon, Tokyo 1995, 195-198. 3. Buyse L. et al.: Collodion baby dehydration: the danger of high transepidermal water loss. Br. J. Dermatol. 1993, 129:86-88. 4. Cambiaghi S., Ermacora E.: Antibiotic therapy in a boy affected by generalized epidermolytic hyperkeratosis. Dermatology 1992, 184:226. 5. Capetanakis J. et al.: Ichthyosis hystrix of „porcupine man” type. Dermatolog. 1975, 151:177-183. 6. Dale B.A., Kam E.: Harlequin ichthyosis. Variability in expression and hypothesis for disease mechanism. Arch. Dermatol. 1993, 129:1471-1477. 7. Epstein E.H.Jr, Leventhal M.E.: Steroid sulfatase of human leukocytes and epidermis and the diagnosis of X-linked ichthyosis. J. Clin. Invest. 1981, 67:1257-1262. 8. Frenk E., de-Techtermann F.: Self-healing collodion baby: evidence for autosomal recessive inheritance. Pediatr. Dermatol. 1992, 9:95-97. 9. Giam Y.C.: Etretinate treatment for harlequin baby. J. Singapore Paediatr. Soc. 1992, 34:217-219. 10. Haftek M. et al.: Modified epiermal lipid composition in air-exposed culture of non-bullous congenital ichthyotic erythroderma (NBCIE) kerationocytes. Arch. Dermatol., 1993, 285:211-215. 11. Haftek M. et al.: A longitudinal study of a harlequin infant evolving towards non-bullous congenital ichthyosiform erythroderma (NBCIE). European Society for Dermatological Research (ESDR), 24th Annual Meeting, Vienna 1994, 198 (abstr.). 12. Hashimoto K., Khan S.: Harlequin fetus with abnormal lamellar granules and giant mitochondria. J. Cutan Pathol. 1992, 19:247-252. 13. Hazell M., Marks R.: Clinical, histologic and cell kinetic discriminants between lumellar ichthyosis and nonbullous congenital ichthyosiform erythroderma. Arch. Dermatol. 1985, 121:489-493. 14. Hoyer H. et al.: Ichthyosis of steroid sulphatase defciency: Clinical study of 76 cases. Dermatologica 1986, 172:184-190. 15. Hubaer M. et al.: Mutations of kerationocyte in transglutaminase lamellar ichthyosis. Science, 1995, 267:525-528. 16. Jacobi H., Heidelbach U.: Erythrodermia congenitalis ichthyosiformis bullosa. Dermatol. Monatschr. 1977, 163:840-843. 17. Langer K. et al.: Kollodiumbaby mit übergang in milde lamellare Ichthyose. Klinischer herlauf, Histopathologie und ultrastrukturalle Befunde. Hautarzt, 1991, 42:34-38. 18. Larregue M. et al.: Le bebe collodion. Evolution a propos de 29 cas. Ann. Dermatol. Syphiligr. 1976, 103:31-56. 19. Larregue M. et al.: Bebe collodion. Trente-deux nouvelles observations. Ann. Dermatol. Venereol. 1986, 113:773-785. 20. Lentz C.L., Altman J.: Lamellar ichthyosis: The natural clinical course of collodion baby. Arch. Dermatol. 1968, 97:3-13. 21. Marghescu S. et al.: Kongenitale reticulare ichthyosiforme erythrodermie. Hautarzt 1984, 35:522-529. 22. Michałowski R. et al.: Netherton-Syndrom mit Alopecie und Prolinurie. Hautarzt 1978, 29:205-208. 23. Milner M.E. et al.: Abnormal lamellar granules in harlequin ichthyosis. J. Invest. Dermatol. 1992, 99:824-829. 24. Nicol A. et al.: Bebe collodion et erythrodermie ichthyosiforme seche. Ann. Dermatol. Venereol. 1986, 113: 147-151. 25. Nychay S.G. et al.: Epiermolytic hyperkeratosis treated with etretinate. Cutis 1991, 47:277-280. 26. Okano M. et al.: X linked ichthyosis and ichthyosis vulgaris: Comparison of thir clinical features based on biochemical analysis. Br. J. Dermatol. 1988, 119:777-783. 27. Pongprasit P.: Collodion baby: The out-come of long-term follow-up. J. Med. Assoc. Thai. 1993, 76:17-22. 28. Prasad R.S. et al.: Menagement and follow-up of harlequin sibilings. Br. J. Dermatol. 1994, 130:650-653. 29. Pulkkinen L. et al.: Epidermolytic hyperkeratosis (bullous congenital ichthyosiform erythroderma). Genetic linkage to chromosome 12q in the region of the type 11 keratin gene cluster. J. Clin. Invest. 1993, 91: 357-361. 30. Rassner G., Steinert U.: Dermatologia: Podręcznik i atlas. Wyd. 1 (polskie), Szubert A. (red.), Urban-Partner; Wrocław 1994, 23-26. 31. Rothangel J.A. et al.: Mutations in the rod domains of keratin 1 and 10 in epidermolytic hyperkeratosis. Science 1992, 257:1128-1130. 32. Rufli T. et al.: Nichtbullose erythrodermie (congenitale) ichthyosiforme mit perinuklearen schalen. Hautarzt 1990, 41:442-447. 33. Russel L.J. et al.: Linkage of autosomal recessive lamellar ichthyosis to chromosome 14q. Am. J. Hum. Genet., 1994, 55:146-152. 34. Shapiro L.J.: Steroid sulfatase deficiency and the genetics of the short arm of the human X chromosome. Adv. Human Genet. 1985, 14:331-381. 35. Snyder A.J. et al.: Genetic mutations in the K1 and K10 genes of patients with epidermolytic hyperkeratosis? Correlation between location and disease severity. J. Clin. Invest. 1994, 93:1533-1542. 36. Terlikowska A. et al.: Brak ekspresji enzymu transglutaminazy-1 w naskórku u rodzeństwa z rybią łuską blaszkowatą. Przegl. Dermatol. 2000, 87:225-232. 37. Unamuno P. et al.: Harlequin foetus on four sibilings. Br. J. Dermatol. 1987, 116: 569-572. 38. Urban J.: Obraz kliniczny zespołu Netherthona z dominującymi objawami atopowymi u 16-miesięcznego dziecka – postępowanie diagnostyczne i lecznicze. XXVI Zjazd PTD, 24-27.09.1998 r., Warszawa, Program prezentacji przypadków, 39 (abstr.). 39. Urban J.: Syndroma Tay (Trichothiodystrophia cum ichthyosis congenita) u 15-leniej dziewczynki. IV Sympozjum Sekcji Derm. Dziec. PTD, Jurata, 28-29 maja 1999 r. Dermatologia kliniczna i zabiegowa 1999, 1 (suppl. 1), 48. 40. Urban J.: Nadmierne rogowacenie dłoni i podeszew – odmiany idiopatyczne oraz w rybiej łusce wrodzonej. IV Sympozjum Derm. Dziec. PTD, Jurata, 28-29 maja 1999 r. Dermatologia kliniczna i zabiegowa 1999, 1 (suppl. 1), 45. 41. Urban J.: Genodermatozy związane z zaburzonym rogowaceniem. [w:] Dermatologia pediatryczna. Miklaszewska M., Wąsik F. (red.), t. 2, Volumed, Wrocław 2000, 297-432. 42. Urban J. et al.: Erythrokeratodermia figurata variabilis. XXVI Zjazd PTD, 24-27.09.1988 r., Warszawa. Program prezentacji przypadków, 43 (abstr.). 43. Urban J. et al.: Keratosis hereditaria extremitatum progrediens Greither. XXVI Zjazd PTD, 24-27.09.1998 r., Warszawa. Program prezentacji przypadków, 74 (abstr.). 44. Urban J. et al.: Mal de Meleda (Keratosis hereditaria transgrediens et progrediens) cum pili monilliformes. XXVI Zjazd PTD, 24-27.09.1998 r., Warszawa. Program prezentacji przypadków, 74 (abstr.). 45. Urban J. et al.: Zespół dziecka kolodionowego (syndroma collodion baby) z przejściem w łagodną postać rybiej łuski blaszkowatej klasycznej (ichthyosis lamellaris classica). XV Ogólnopolskie Sympozjum Neonatologiczne, Poznań, 28-30.09.2000 r. Postępy w Neonatologii, t. XI (w druku). 46. Wells R.S., Jenning M.C.: X Linked ichthyosis and ichthyosis vulgaris: clinical and genetic distinctions in a second series of families. J.A.M.A., 1978, 93, suppl., 59-60. 47. Williams M.L., Elias P.M.: Heterogenity in autosomal recessive ichthyosis. Clinical and biochemical differentation of lamellar ichthyosis and nonbullous congenital ichthyosiform erythroderma. Arch. Dermatol., 1985, 121:477-488.