Izabela Korczowska, Jan K. Łącki
Etiopatogeneza osteoporozy
pathogenesis of osteoporosis
z Kliniki Reumatologii i Immunologii Klinicznej AM w Poznaniu
Kierownik Kliniki: prof. dr hab. med Jan K. Łącki
Streszczenie
Osteoporosis is defined as a systemic skeletal disorder characterised by low bone mass and microarchitectural deterioration of bone tissue, with a consequent increase susceptibility to fracture. Most simply osteoporosis arises from an imbalance of bone formation and bone resorption. After the explosion of information on risk factors produced recently our knowledge on osteoporosis has been significantly improved, however it seems that it still is a puzzle for us. The final understanding of processes underlying the osteoporosis pathogenesis is still crucial for the most efficient and safe therapy.

BUDOWA KOŚCI
Osteoporoza jest systemową chorobą charakteryzującą się obniżeniem masy kostnej i zaburzeniami mikroarchitektury tkanki kostnej, czego konsekwencją jest mniejsza wytrzymałość kości, która w następstwie nawet niedużego urazu może ulec złamaniu. Aby zrozumieć patogenezę warto przypomnieć prawidłową anatomię i fizjologię kości. Szkielet dorosłego człowieka tworzy kość korowa (zbita) oraz kość beleczkowa (gąbczasta). Kość korowa obecna jest głównie w trzonach kości długich i tworzy powierzchnię kości płaskich. Zbudowana jest z gęstej tkanki kostnej położonej koncentrycznie wokół kanałów Haversa, zawierających naczynia krwionośne i limfatyczne, nerwy i tkankę łączną. Kość beleczkowa występująca głównie na końcach kości długich, wewnątrz kości płaskich, składa się z łączących się blaszek i beleczek (1). Chociaż kość korowa różni się strukturą od kości beleczkowej, mają one taką samą budowę molekularną i biochemiczną. Kość składa się z komórek oraz zewnątrzkomórkowej macierzy (ECM). ECM posiada komponentę mineralną i niemineralną. Ta ostatnia zwana osteoidem, jest produkowana i wydzielana przez osteoblasty. Osteoid zbudowany jest z kolagenu typu I oraz białek niekolagenowych (osteokalcyna, białko macierzy GLA, osteonektyna i inne) Komponent mineralny stanowią wapniowe hydroksyapatyty oraz jony fosforanowe (1).
Choć zarówno kość beleczkowa jak i korowa odgrywają ważną rolę w mechanicznych właściwościach szkieletu, ich podatność na osteoporozę jest odmienna. Cechą morfologiczną odróżniającą ostoporozę posteroidową od pomenopauzalnej jest odmienny typ zmniejszenia liczby i grubości beleczek. W osteoporozie pomenopauzalnej odnotowano 45% spadek liczby beleczek i 5% ścieczenie, w posteroidowej zaś tylko 10% spadek liczby beleczek i aż 30% zwężenie. W przypadku kości gąbczastej beleczki mogą tracić prawidłową grubość lub może nastąpić penetracja i erozja. Procesowi zwężenia beleczek towarzyszy lepsze zachowanie architektury kości niż w przypadku penetracji i erozji charakterystycznej dla osteoporozy pomenopauzalnej (1).
Komórki kości to przede wszystkim osteoblasty, osteocyty i osteoklasty. Osteoblasty, komórki pochodzące od wielopotencjalnych komórek macierzystych zrębu, odpowiedzialne są za tworzenie kości, wydzielają osteoid oraz doprowadzają do mineralizacji macierzy kostnej (2). Włókna kolagenowe znajdujące się wewnątrz osteoidu ułożone są w liniowe kolumny tworząc pory i jamki, i w tych właśnie miejscach zainicjowana zostaje mineralizacja. Osteoblasty posiadają receptory dla wielu czynników wpływających na metabolizm kości (najważniejsze z nich to hormon przytarczyc – PTH oraz 1,25-dihydroxy-witamina D). Osteoblasty odgrywają dużą rolę w różnicowaniu i aktywowaniu osteoklastów.
Osteoklasty, to duże wielojądrzaste komórki pochodzące z prekursorów hematopoezy należących do rodziny monocytów-makrofagów, odpowiedzialne za resorpcję kości. Osteoklast łączy się z macierzą kostną za pośrednictwem integryn, tworząc jamkę resorpcyjną (2), do której uwalnia proteazy, włączając różne katepsyny oraz metaloproteinazy (MMPs), powodujące niszczenie macierzy kostnej.
Integryny są białkami adhezyjnymi obecnymi na powierzchni błon cytoplazmatycznych, zbudowane z łańcuchów a i b. Integryny wiążą się z kością przez sekwencje RGD (Arg-Gly-Asp), które wchodzą w skład białek wiążących: vitonektyny, ostoponiny i sialoprotein kostnych II. Białka te są rozpoznawane głównie przez rodzinę av integryn, głównie avb3 oraz avb5, te ostatnie uznane zostały za marker prekursora osteoklastów, a avb3 dojrzałych osteoklastów (2). Badania na szczurach wykazały, że zablokowanie integryn avb3 powoduje zahamowanie resorpcji kości. Inoue i wsp. (3) stwierdzili, że różne cytokiny (m.in. IL-4, TNF-alfa) oraz 1,25(OH)2D3 modulują ekspresję avb3 oraz avb5. Proces resorpcji zachodzący wewnątrz osteoklasta jest zainicjowany przez przyłączenie cząsteczki wody do dwutlenku węgla. W wyniku tego procesu powstaje kwas węglowy, który następnie dysocjuje do jonu wodoru oraz jonu HCO3-. Jony wodorowe pompowane są przez rąbek szczoteczkowy i pompę protonową (vacuolar ATP-asa) do jamki resorpcyjnej. Jony wodorowęglanowe są wymieniane na jony Cl- w części nieresorpcyjnej. Jony chloru są następnie wydzielane do części pomiędzy osteoblastem a kością i wraz z jonami wodorowymi powodują zakwaszenie środowiska do pH ok. 4,5 (ryc. 1). To wysoko kwaśne mikrośrodowisko hamuje mineralizację kości (2). Pompa protonowa odgrywa ważną rolę w prawidłowym działaniu osteoklastów, jej mutacja może być przyczyną osteopetrozy (4). Początkowo aktywność pompy protonowej wiązano z osteoklastem, ale badania kilku ostatnich lat wykazały, że wiąże się również z osteoblastem, aktywowanym przez hormony przytarczyc. W ten sposób osteoblasty będąc dodatkowym źródłem jonów wodorowych w tkance kostnej, mogą przyczyniać się do degradacji kości (5). Osteoklasty nie posiadają receptorów dla PTH oraz 1,25(OH)2D3 (1), jednakże związki te wpływają na aktywność osteoklastów poprzez stymulację osteoblastów.

Ryc. 1. Mechanizm resorpcji tkanki kostnej.
Osteocyty są małymi płaskimi komórkami wewnątrz macierzy kostnej. Odgrywają ważną rolę w odpowiedzi tkanki kostnej na stymulację mechaniczną. W kości gąbczastej ułożone są równolegle do włókien kolagenu, zaś w kości korowej uporządkowane są obwodowo wokół koncentrycznych blaszek kostnych (1).
PROCES PRZEBUDOWY TKANKI KOSTNEJ
Całkowity proces przebudowy tkanki kostnej u dorosłego człowieka trwa około 3-6 miesięcy. Pierwszym jej etapem jest aktywacja (przemiana określonego fragmentu powierzchni kości ze stanu spoczynku do aktywności). Przebudowa rozpoczyna się od cofnięcia komórek wyściółki i usunięcia cienkiej błony kolagenowej pokrywającej powierzchnię mineralizowanej kości. Kolejno dochodzi do miejscowego gromadzenia się i aktywacji osteoklastów, które resorbują kość za pomocą wydzielanej mieszaniny kwaśnych i obojętnych enzymów proteolitycznych (ryc. 2). Następnie w miejsce wytworzonej jamki migrują jednojądrowe komórki, które odkładają tzw. linię cementową, będącą granicą nowego osteoidu. Dzięki aktywności osteoblastów dochodzi do produkcji osteoidu i wypełnienia jamki resorpcyjnej nowo zsyntetyzowaną kością. Ostatnim etapem jest mineralizacja nowo powstałej tkanki kostnej. Część osteoblastów zostaje wykorzystana do syntezy osteoidu (przekształca się w osteocyty), pozostałe zaś znajdują się na powierzchni kości w postaci komórek osteogennych (wyściełających). Aktywność osteoklastów jest wielokrotnie wyższa niż osteoblastów. Ocenia się, że do wypełnienia ubytku wytworzonego przez jeden osteoklast potrzebnych jest około 200 osteoblastów. Stała przebudowa tkanki kostnej ułatwia procesy naprawcze i adaptacyjne.
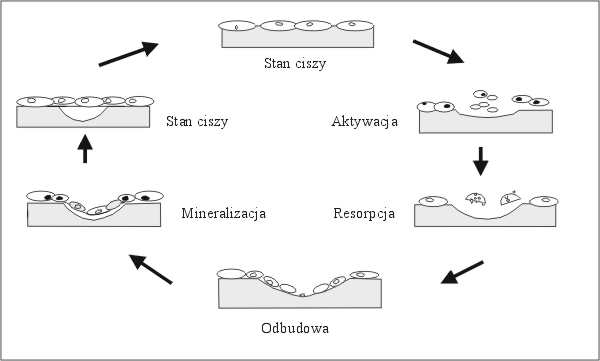
Ryc. 2. Proces przebudowy tkanki kostnej.

Ryc. 3. Najważniejsze czynniki wpływające na procesy tworzenia i resorpcji kości.
UTRATA MASY KOSTNEJ
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Bono C.M., Einhorn T.A.: Overview of osteoporosis: pathophysiology and determinants of bone strength. Eur. Spine J. 2003, 12:S90-S96. 2. Teitelbaum S.L.: Osteoclasts, integrins and osteoporosis. J. Bone Miner. Metab. 2000, 18:344-349. 3. Inoue M. et al: Tumor necrosis factor a regulates avb5 integrin expression by osteoclast precursors in vitro and in vivo. Endocrinology 2000, 141:284-290. 4. Kornak U. et al: Mutation in the a3 subunit of the vacuolar H(+)-ATP-ase cause infantile malignant osteopetrosis. Hum. Mol. Genet. 2000, 9:2059-2063. 5. Barret M.G. et al: A new action of parathyroid hormone. Receptor-mediated stimulation of extracellular acidification in human osteoblast-like SaOS-2 cells. J. Biol. Chem. 1997, 272:26346-26353. 6. Ralston S.H. et al: Nitric oxide: a cytokine-induced regulator of bone resorption. J. Bone Miner. Res. 1995, 10:1040-1049. 7. Steinbeck M.J. et al: NADPH-oxidase expression and in situ production of superoxide by osteoclasts actively resorbing bone. J. Cell Biol. 1994, 126:765-772. 8. Eisman J.A.: Genetics of osteoporosis. Endocr. Rev 1999, 20:788-804. 9. Ferrari S.L. et al: Bone mineral mass and calcium and phosphate metabolism in young men: relationship with vitamin D receptor allelic polymorphism. J. Clin. Endocrinol. Metab. 1999, 84:2043-2048. 10. Lorentzon M.: Vitamin D receptor gene polymorphism is associated with birth height, growth to adolescence and adult stature in healthy Caucasian men: a cross-sectional and longitudinal study. J. Clin. Endocrinol Metab 2000, 85:1666-1670. 11. Langdahl B.L. et al: Polymorphism in the vitamin D receptor gene and bone mass, bone turnover and osteoporotic fractures. Eur. J. Clin. Invest. 2000, 30:608-617. 12. Spotila L.D. et al: Mutation analysis of coding sequences for type I procollagen I individuals with low bone density. J. Bone Miner. Res. 9:923-932. 13. Keen R.W. et al: Associated of polymorphism at the type I collagen (COLIA1) locus with reduced bone mineral density, increased fracture risk and increased collagen turnover. Arthritis Rheum. 1999, 42:285-290. 14. Kann P. et al: The collagen I a1 SP1 polymorphism is associated with differences in ultrasound transmission velocity in the calcaneus in postmenopausal women. Calcif Tissue Int. 2002, 70:450-456. 15. Grant S.F.A. et al: Reduced bone density and osteoporosis associated with polymorphic SP1 site in the collagen type I alpha 1 gene. Nat. Genet 1996, 14:203-205. 16. Uitterlinden A.G. et al: Relation of alleles of the collagen type Ia1 gene to bone density and risk of osteoporotic fractures in postmenopusal women. N. Engl. J. Med. 1998, 338:1016-1021. 17. Mann V. et al: A COLIA1 Spl binding site polymorphism predisposes to osteoporotic fracture by affecting bone density and quality. J. Clin. Invest 2001, 107:899-907. 18. Ashford R.U. et al: Studies of bone density, quantitative ultrasound and vertebral fractures in relation to collagen type I alpha 1 alleles in elderly women. Calcif Tissue Int. 2001, 68:348-351. 19. Pluijm S.M.F. et al: Collagen type I a Sp1 polymorphism, osteoporosis and intervertebral disc degeneration in older men and women. Ann. Rheum. Dis. 2004, 63:71-77. 20. Gennari L., Brandi M.L.: Genetics of male osteoporosis. Calcif. Tissue Int. 2001, 69:200-204. 21. Rosen C.J. et al: Association between serum insulin growth factor–I (IGF-I) and a simple sequence repeat in IGF-I gene: implications for genetic studies of bone mineral density. J. Clin. Endocrinol Metab. 1998, 83:2286-2290. 22. Takacs I. et al: Sibiling pair linkage and association studies between bone mineral density and the insulin-like growth factor I gene locus. J. Clin. Endocrinol. Metab. 1999, 84:4467-4471. 23. Braga V. et al: Relationship among VDR (BsmI and FokI), COLIA1, and CTR polymorphisms with bone mass, bone turnover markers, and sex hormones in men. Calcif Tissue Int. 2002, 70:457-462. 24. Ralston S.H. et al: Analysis of gene expression in human bone biopsies by polymerase chain reaction: evidence for enhanced cytokine expression in postmenopausal osteoporosis. J. Bone Miner. Res. 1994, 9:883-890. 25. Ota N. et al: A nucleotide variant in promoter region of the inerleukin-6 gene associated with decreased bone mineral density. J. Hum. Genet. 2001, 46:267-272.