Danuta Maślińska
Programowana śmierć komórki (apoptoza)
w procesie zapalnym
Programmed cell death (apoptosis) in inflammation
z Zakładu Neuropatologii Rozwojowej Instytutu Centrum Medycyny Doświadczalnej i Klinicznej
Polskiej Akademii Nauk w Warszawie i Katedry Patologii Ogólnej i Doświadczalnej
Akademii Medycznej w Warszawie
Kierownik Zakładu: doc. dr hab. med. Danuta Maślińska

Organizmy wielokomórkowe narażone na stałe oddziaływanie środowiskowych czynników uszkadzających (infekcyjnych, toksycznych, jonizujących itp.) wykształciły podczas ewolucji złożony system mechanizmów obronnych, do których należy proces zapalny. Istotną rolę w indukcji i modulacji tego procesu odgrywa śmierć komórki. Wyróżnia się dwa rodzaje śmierci komórki: martwicę (nekrozę) i apoptozę (programowaną śmierć).
Martwica jest śmiercią wywołaną silnym działaniem dowolnego czynnika uszkadzającego, który przerywa w komórce, podstawowe procesy życiowe (oddychanie, produkcję związków wysokoenergetycznych). Szybko dochodzi do obrzęku, rozpadu organelli, uszkodzenia błony komórkowej. Zawartość komórki wylewa się do przestrzeni pozakomórkowej, niszcząc macierz i uszkadzając sąsiednie komórki. Powstaje ognisko martwicy, do którego w ramach reakcji obronnej napływają granulocyty obojętnochłonne, limfocyty i makrofagi biorąc udział w procesie zapalnym. Zadaniem tych komórek jest usunięcie mikroorganizmów, ciał obcych i martwych tkanek oraz stworzenie warunków do regeneracji i bliznowacenia. Aby spełnić swą rolę, komórki produkują i uwalniają wiele substancji o silnym działaniu destrukcyjnym i prozapalnym. Dlatego liczba tych komórek i czas ich pobytu w ognisku zapalnym muszą być ściśle nadzorowane. Sprawne i bezpieczne likwidowanie komórek, po wykonaniu przez nie wyznaczonego zadania, pozwala uniknąć przedłużania się procesu zapalnego i uszkodzenia zdrowych komórek i tkanek. Uniwersalnym sposobem usuwania komórek zapalnych jest ich samozagłada, czyli apoptoza lub programowana śmierć. W odróżnieniu od martwicy nie jest to śmierć przypadkowa. Ulegają jej tylko te komórki, które mogą odebrać „rozkaz śmierci”. Mają one na swojej powierzchni białkowe struktury receptorowe ? „receptory śmierci”, które łączą się ze swoistym białkiem (ligandem) produkowanym przez komórki przesyłające sygnał apoptozy (29). Najlepiej dotychczas poznano receptory należące do rodziny receptorów czynnika martwicy nowotworów (Tumor necrosis factor receptor ? TNFR) (ryc. 1).
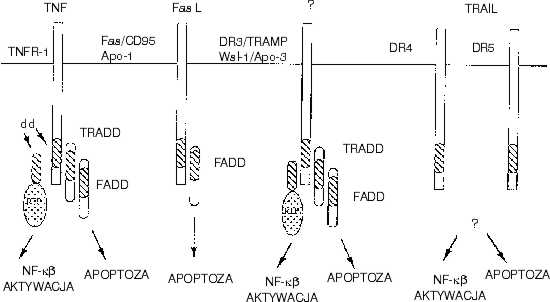
Receptory śmierci (TNFR-1, Fas/CD 95/Apo-1, DR3/TRAMP, DR4, DR5); swoiste ligandy (TNF, Fas L, TRAIL); białka uczestniczące w przekazywaniu sygnału apoptozy (TRADD, FADD), domeny śmierci (dd); czynnik jądrowy (NK-kb). Według: M. Muzio, Int. J. Clin. Lab. Res., 28:141-147, 1998.
Ryc. 1. Receptory i białka uczestniczące w przekazywaniu sygnału apoptozy.
Receptory te charakteryzuje bogata zawartość cysteiny w zewnątrzkomórkowej części białka oraz obecność w wewnątrzkomórkowej części białka receptorowego regionu niezbędnego do przekazywania sygnału apoptozy, tzw. domeny śmierci (death domain). Obecność tej domeny pozwala receptorowi na interakcję z kompletem swoistych białek komórkowych (adapters) zdolnych przekazać sygnał do struktur efektorowych komórki (11, 23, 31, 32, 40). Każdy z „receptorów śmierci” należący do grupy TNFR ma inne wymagania odnośnie do interakcji z wewnątrzkomórkowymi białkami uczestniczącymi w przekazywaniu sygnału. Poza poznanym receptorowym przekazem, istnieje szereg innych mało poznanych sposobów indukcji apoptozy. Jednym z nich jest penetracja do komórki granzymu B ? substancji uwalnianej z ziarnistości cytoplazmatycznych limfocytów cytotoksycznych (37). Istnieje więc, prawdopodobnie szereg dróg transdukcji otrzymanego sygnału apoptozy. Być może, że każdy z tych układów jest charakterystyczny dla określonego typu komórki lub, że w jednej komórce istnieje kilka możliwości przekazania odebranego sygnału. Transdukcja sygnału apoptozy w komórce jest zjawiskiem złożonym i wymaga dostarczenia znacznej ilości energii. Komórka uczestniczy czynnie we własnej samozagładzie, przystosowując metabolizm do wymagań toczących się w niej przemian. Egzekutorami sygnału śmierci są enzymy cytosolowe ? kaspazy. W zdrowych komórkach występują w postaci zymogenów. Indukcja apoptozy polega na oderwaniu od zymogenu fragmentu białka blokującego aktywność enzymu. Pierwszą kaspazą zidentyfikowaną w komórkach ssaków był enzym konwertujący interleukinę-1b (interleukin-1b converting enzyme, ICE). Dalsze badania wykazały obecność wielu kaspaz podobnych do ICE. Wszystkie poznane kaspazy charakteryzują się tym, że przecinają łańcuch białkowy substratu w miejscu obecności w nim kwasu asparaginowego, oraz że same dla siebie są enzymami aktywującymi. W związku z tym, aktywacja jednej z kaspaz może wywołać kaskadową reakcję, podczas której uwalnia się z zymogenów wiele aktywnych kaspaz. Kaspazy niszczą białka enzymatyczne i strukturalne komórki. Degradacja białkowej kinazy DNA i polimerazy poli ADP rybozy (PARP) uniemożliwia naprawę uszkodzonego DNA (7). Zniszczenie laminy uszkadza błonę jądrową (39), gelsoliny, aktyny i filamentów pośrednich cytoskeletonu ? zmienia kształt komórki (6, 25, 30). Łańcuch DNA zostaje pocięty na krótkie fragmenty przez endonukleazę CAD (caspase-activated DNase). Sama nukleaza nie jest substratem kaspaz, natomiast ulega aktywacji po zniszczeniu przez kaspazy inhibitora, który w normalnych warunkach blokuje CAD (14, 36). Poznano dotychczas 25 kaspaz, ale najważniejszą rolę w procesie apoptozy odgrywa kaspaza 9. Wchodzi ona w skład kompleksu, który nazwano apoptosomem (9). Apoptosom składa się poza kaspazą 9 z czynnika 1 aktywującego kaspazy (apoptotic protease activity factor-1, Apaf-1) i z białek blokujących proces apoptozy bcl-2/xL (ryc. 2). Funkcja apoptosomu jest modulowana przez szereg białek, takich jak proapoptotyczne białka bad, bik i bid, które również należą do rodziny bcl-2. Wiążąc się z białkami bcl-2/xL białka bad, bik i bid ułatwiają aktywację kaspazy 9 przez Apaf-1 (10, 19). Interakcja trzech składowych apoptosomu decyzuje o przeżyciu lub śmierci komórki (9).
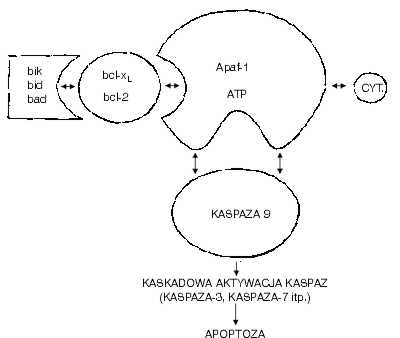
Norma: Interakcja białek bcl-2 i bcl-xL w Apaf-1 zapobiega aktywacji kaspazy 9. Apoptoza: Interakcja białek bid, bik i bad z białkami bcl-2 i bclxL ułatwia aktywację kaspazy 9 przez Apaf-1, prowadząc do kaskadowej aktywacji innych kaspaz i apoptozy. Białka działające proapoptotycznie: bik, bid i bad; białka blokujące apoptozę: bcl-2, bcl-xL; czynnik aktywujący kaspazę: Apaf-1; kofaktor apoptosomu: Cyt. cytochrom C. Według: A.M. Chinnaiyan, Neoplasia 1:5-15, 1999.
Ryc. 2. Apoptosom ssaków.
Podczas apoptozy szereg nowych genów ulega indukcji. Obserwuje się syntezę białek czynników transkrypcyjnych c-fos i c-jun, które tworząc dimer AP-1 przyspieszają odczyt informacji z indukowanych genów. W cytoplazmie komórki pojawia się wiele nowych białek o właściwościach enzymatycznych pogłębiając chaos metaboliczny w umierającej komórce. Komórka obkurcza się i zapada, jak namiot, w którym przecinane są coraz to nowe linki utrzymujące jego kształt. Mimo to, w przeciwieństwie do martwicy, podczas apoptozy komórka zachowuje integralność swoich organelli. Powierzchnia obkurczającej się komórki pokrywa się licznymi drobnymi uwypukleniami (35). Błona komórkowa ulega przekształceniom strukturalnym. Zmiany te polegają między innymi na
przemieszczeniu się fosfatydyloseryny z wewnętrznej do zewnętrznej warstwy błony komórkowej (33). Zmiana ta ma szczególne znaczenie, ponieważ ułatwia leżącym w sąsiedztwie komórkom szybkie rozpoznanie, fagocytowanie, a następnie trawienie materiału apoptotycznego. Komórka apoptotyczna może być połknięta w całości lub rozpaść się na szereg obłonionych fragmentów (ciał apoptotycznych) zawierających skondensowane masy jądra i cytoplazmy. Ciała apoptotyczne są również szybko usuwane na drodze fagocytozy. Ten typ fagocytozy nie wymaga obecności aktywnych makrofagów, które produkując substancje prozapalne, destrukcyjnie działające na tkankę, mogłyby inicjować proces zapalny. Niezależnie od liczby komórek ulegających apoptozie, każda z nich umiera indywidualnie, tzn. że nie wywiera wpływu na podobny proces zachodzący w innych komórkach. W związku z tym nie powstaje ogniskowe uszkodzenie tkanki, ubytek który wymagałby tworzenia się blizny. Komórki apoptotyczne znikają, nie pozostawiając po sobie śladu i nie wywołując odczynu zapalnego.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Alcami A., Smith G.L.: Cytokine receptors encodes by poxviruses: a lesson in cytokine biology. Immunol. Today 1995, 16:474. 2. Arend W.P., Dayer J-M.: Cytokines and cytokine inhibitors or antagonists in rheumatoid arthritis. Arthritis Rheum. 1990, 33:305. 3. Baum J. et al.: Laboratory findings in rheumatoid arthritis [W:] McCarty D.J. Koppman W.J., Red. Arthritis and allied conditions, a textbook of rheumatology, 12th edn. Philadelphia: Lea & Febiger, 1993, 841. 4. Bertin J. et al.: Death effector domin-containing herpesvirus and poxvirus proteins inhibit both Fas and TNFR 1 ? induced apoptosis. Proc. Natl. Acad. Sci. USA, 1997, 94:1172. 5. Bump N.J. et al.: Inhibition of ICE family proteases by baculovirus antiapoptotic protein p53. Science. 1995, 269:1885. 6. Caulin C. et al.: Caspase cleavage of kreatin 18 and reorganization of intermediate filaments during epithelial cell apoptosis. J. Cell Biol. 1997, 138:1379. 7. Casciola-Roseen L. et al.: DNA ? depend protein kinase is one of a subset of autoantigens specifically cleaved early during apoptosis. J. Exp. Med. 1995, 182:1625. 8. Cheng EH-Y. et al.: Conversion of Bcl-2 a Bax-like death effector by caspases. Science 1997, 278:1966. 9. Chinnaiyan A.M.: The apoptosome: Heart and soul of the cell death machine. Neoplasia 1999, 1:5. 10. Chinnaiyan A.M. et al.: Interaction of CED-4 with CED-3 and CED-9: a molecular framework for cell death. Science 1997, 275:1122. 11. Chinnaiyan A.M. et al.: Signal transduction by DR3, a death doomain-containing receptor related to TNFR-1 and CD95. Science 1996, 274-990. 12. Chu CQ et al.: Localization of tumor necrosis factor a in synovial tissues and at the cartilage-pannus junction in patients with rheumatoid arthritis. Arthritis Rheum. 1991, 34:1125. 13. Deitch E.A. et al.: Hypertrophic scars: Analysis of variables. J. Trauma 1983, 23:895. 14. Enari M. et al.: A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor, ICAD. Nature, 1997, 39:43. 15. Feldmann M. et al.: Rheumatoid arthritis. Cell 1996, 85:307. 16. Firestein G.S. et al.: Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 1997, 94:10895. 17. Greenhalgh D.G.: The role of apoptosis in wound healing. Int. J. Biochem & Cell Biol., 1998, 30:1019. 18. Harris E.D.Jr: Mechanisms of disease: rheumatoid arthritis: pathophysiology and implications for therapy. N. Engl. J.Med., 1990, 322:1277. 19. Hawkins C.J. et al.: Inhibition of interleukin 1 beta-converting enzyme-mediated apoptosis of mammalin cells by baculovirus IAP. Proc. Natl. Acad. Sci. USA, 1996, 93:13786. 20. Hengartner M.O., Horovitz H.R.: C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell 1994, 76:665. 21. Hu S. et al.: A novel family of viral death effector domain-containing molecules that inhibit both CD-95 and tumor necrosis factor receptor-1 ? induced apoptosis. J. Biol. Chem. 1997, 272:9621. 22. Hunt T.K. et al.: Studies on inflammation and wound healing. Angiogenesis and collagen synthesis stimulated in vivo by resident and activated wound macrophages. Surgery, 1984, 96:48. 23. Itoh N., Nagata S.: A novel protein domain required for apoptosis. Multinational analysis of human Fas antigen. J. Biol. Chem. 1993, 268:10932. 24. Irmler M. et al.: Inhibition of death receptor signals by cellular FLIP. Nature 1997, 388:190. 25. Kothakota S. et al.: Caspase-3-generated fragment of gelsolin effector of morphological changes in apoptosis. Science 1997, 278:294. 26. Levine A.J.: p53, the cellular gatekeeper for growth and division. Cell 1997, 88:323. 27. Meyer F.A. et al.: Synergistic additive and antagonistic effects of interleukin-1b, tumor necrosis factor a and g-interferon on prostaglandin E, hyaluronic acid, and collagenase production by cultured synovial fibroblasts. Arthritis Rheum. 1990, 33:1518. 28. Muller-Ladner U.: T cell-independent cellular pathways of rheumatoid joint destruction. Curr. Opi. Rheumatol. 1995, 7:222. 29. Muzio M.: Signalling by proteolysis: death receptors induce apoptosis. Int. J. Clin. Lab. Res. 1998, 1998, 28:141. 30. Na S. et al.: D4-GDI, a substrate of CPP32 is proteolyzed during Fas-induced apoptosis. J. Biol. Chem. 1996, 271:1109. 31. Pan G. et al.: The receptor for the cytotoxic lignad TRAIL. Science, 1997, 276:111. 32. Pan G. et al.: An antagonist decoy receptor and a seath domain-containing receptor for TRAIL. Science 1997, 277:815. 33. Pittoni V., Isenberg D.: Apoptosis and antiphospholipid antibodies. Semin. Arthritis Rheum. 1998, 28:163. 34. Roberts A.B., Sporn M.B.: Physiological actions and clinical applications of transforming growth factor-b (TGF-b) Growth Factors. 1993, 8:1. 35. Rosen A., Casiola-Rosen L.: Autoantigens as substrates for apoptotic proteases: implications for the pathogenesis of systemic autoimmune disease. Cell death and differentiation. 1996, 6:6. 36. Sakahira H. Et al.: Cleavage of CDA inhibitor in CAD activation and DNA degradation during apoptosis. Nature 1997, 39:96. 37. Spaeny-Dekking EHA et al.: Extracellular granzymes A and B in humans: detection of native species during CTL responses in vitro and in vivo. J. Immunol. 1998, 160:3610. 38. 11(6) Stashenko P. et al.: Synergistic interaction between interleukin 1, tumor necrosis factor, and lymphotoxin in bone resorption J. Immunol., 1987, 138:1464. 39. Takahashi A. et al.: Multiple interleukin-1b-converting enzyme-related proteases with distinct recognition properties are active in apoptosis. Proc. Natl. Acad. Sci. USA 1996, 93:8395. 40. Tartaglia L.A. et al.: A novel domain within the 55 kd TNF receptor signals cell death. Cell 1993, 74:845. 41. Tewari M.: Crm A, a poxvirus ? enconded serpin, inhibits cytotoxic T-lymphocyte-mediated apoptosis. J. Biol Chem. 1995, 270:705. 42. Thome M. et al.: Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997, 386:517. 43. Tidball J.G., Pierre B.A.St.: Apoptosis of macrophages during the resolution of muscle inflammation. J. Leukoc. Bid. 1996, 59:380. 44. Wakisaka S. et al.: Modulation by proinflammatory cytokines of Fas/Fas ligand-mediated apoptotic cell death of synovial cells in patients with rheumatoid arthritis (RA). Clin. Exp. Immunol. 1998, 114:119. 45. Yaron I. et al.: Some recombinant human cytokines stimulate glycosaminoglycan synthesis in human synovial fibroblast cultures and inhibit it in human articulat cartilage cultures. Arthritis Rheum. 1989, 32:173-180. 46. Zhou Q et al.: Target protease specificity of the viral serpin CmrA. Analysis of five caspases. J. Biol. Chem. 1997. 272:7797. 47. Zvaifler N.J.: Etiology and pathogenesis of rheumatoid arthritis. [W:] McCarty D.J., Koopman W.J. red. Arthritis and allied conditions a textbook of rheumatology, 12th edn. Philadelphia: Lea & Febiger, 1993, 723-736.