© Borgis - Nowa Pediatria 1/2002, s. 7-11
Danuta Rosińska-Borkowska1, Aldona Ceregra2, Anna Chrupek-Gozdek2
Acrodermatitis Enteropathica w świetle wieloletnich obserwacji*
Long lasting observations of patient with Acrodermatitis Enteropathica
1 z Oddziału Dermatologii Dziecięcej Szpitala im. Św. Łazarza w Warszawie
Ordynator Oddziału: dr n. med. Danuta Rosińska-Borkowska
2 z Poradni Dermatologicznej Kliniki Pediatrii Diabetologii i Alergologii Instytutu Pomnik – Centrum Zdrowia Dziecka w Warszawie
Kierownik Kliniki: prof. dr hab. n. med. Elżbieta Piontek
Streszczenie
The aim of this study was to present our observations of clinical course and the management in 26 patients with Acrodermatitis Enteropathica.
Doses of ZnSO4 were individualy chosen to each patient. The obtained data point to the occurance of improvement in clinical state and reduce of diseases symptoms during adolescence in our patients.

Acrodermatitis Enteropathica (AE) jest rzadko występującą jednostką chorobową manifestującą się objawami skórnymi i zaburzeniami żołądkowo-jelitowymi o ciężkim przebiegu klinicznym. Po raz pierwszy opisali ją w 1932 roku Danbolt i Closs (2), a w 1936 roku Brandt (2). W 1942 roku nazwano ją zespołem Danbolta-Brandta-Clossa (2). Etiopatogeneza tego schorzenia była długo nie znana i nadal budzi kontrowersje (1, 2, 3). Stwierdzono, że jest to choroba genetycznie uwarunkowana, dziedzicząca się autosomalnie recesywnie (9). Występuje jednakowo u obu płci.
Donoszono o jej występowaniu u rodzeństwa (9, 16, 17). Dzięki badaniom Moynahana i Burnesa w latach 1973-1974, nastąpił przełom w poglądach na patogenezę i leczenie AE (10). Autorzy ci (10) stwierdzili, że istotą choroby jest niedobór cynku w surowicy krwi, spowodowany jego upośledzonym wchłanianiem z przewodu pokarmowego. Do dnia dzisiejszego nie ustalono przyczyny upośledzonego wchłaniania cynku. Wyrażano pogląd, że defekt enzymatyczny błony śluzowej jelita cienkiego polega na obniżeniu aktywności enzymów, głównie dehydrogenazy bursztynianowej i aminopeptydazy leucyny (16). Moynahan i Burnes wysunęli przypuszczenie, że powstające w procesie trawienia białek oligopeptydy u chorych z AE chelatują cynk znajdujący się w diecie, tworząc z nim nierozpuszczalne kompleksy (10).
Powstanie toksycznych oligopeptydów związane jest z podejrzeniem wadliwej przemiany tryptofanu u tych chorych (11, 16). Evans i Johnson przedstawili pogląd, że istnieje czynnik wiążący (Zn-binding factor). Jest on wydzielany przez trzustkę do światła przewodu pokarmowego, skąd transportuje związany cynk do komórki błony śluzowej jelita cienkiego (3). Ustalono również, że Zn-binding factor (3) obecny jest w mleku kobiecym, brak go natomiast w mleku krowim. Tłumaczyłoby to fakt, częstego pojawiania się pierwszych objawów typowych dla AE po przejściu z karmienia naturalnego na sztuczne (11, 17). Czynnik ten został zidentyfikowany jako prostaglandyna E2 (9). U pacjentów z AE stwierdzono również niski poziom kwasu arachidonowego. Okazało się, że chorzy z AE nie są w stanie syntetyzować ilości prostaglandyny wystarczającej do utworzenia Zn-binding factor.
Oprócz AE uwarunkowanej genetycznie, opisywane są przypadki z objawami klinicznymi przypominającymi AE (tzw. AE-like). Są to pacjenci z niedostateczną podażą cynku lub nadmierną jego utratą w przebiegu różnych chorób.
Odżywiani parenteralnie, wcześniaki karmione wyłącznie humanizowanym mlekiem, stosowanie diety ubogo białkowej, w chorobach z towarzyszącą przewlekłą biegunką, w zwłóknieniu torbielowatym, w chorobie syropu klonowego, w przypadku choroby Crohna, glukagonoma, u alkoholików (6, 8, 14, 17). Opisywane są również przypadki nabytego niedoboru cynku, u wcześniaków karmionych wyłącznie piersią, u których przyczyną choroby jest niski poziom cynku w mleku matki przy prawidłowym poziomie w surowicy. Prawdopodobnie jest to związane z anomalią przekazywania cynku z osocza do mleka matki (1, 7, 12, 15). Poziom cynku jest więc zbyt mały w stosunku do zwiększonych potrzeb wcześniaka (niedojrzałość układu trawiennego, zmniejszona resorbcja, wzmożone wydalanie jelitowe, małe rezerwy w mięśniach i kościach).
Celem pracy było przedstawienie wieloletnich obserwacji dotyczących przebiegu choroby i leczenia dwudziestu sześciu pacjentów (dzieci i dorośli) z AE oraz przydatność oznaczania poziomu cynku w surowicy i krwinkach w diagnozowaniu i monitorowaniu leczenia.
Materiał i metodyka
Obserwacje dotyczą 26 pacjentów w wieku od 4 miesięcy do 44 rż., którzy byli hospitalizowani a następnie obserwowani w Oddziale i Poradni Dermatologii Dziecięcej Szpitala im. św. Łazarza w Warszawie oraz Poradni Dermatologicznej IP CZD w latach 1968-2000. W grupie 26 pacjentów było troje dzieci w wieku do 2 rż., pięcioro od 2 do 16 rż. i 18 osób dorosłych w wieku powyżej 16 rż. (ryc. 1).
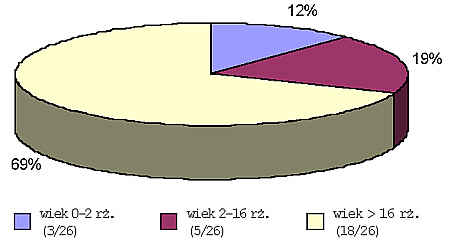
Ryc. 1. Wiek pacjentów.
Tabela 1. Materiał kliniczny.
| Płeć | Ż | M |
| Liczba pacjentów (26) | 15/26 (2 siostry) | 11/26 (3 braci) |
Pośród 11 mężczyzn (chłopców) było trzech braci, a wśród 15 kobiet (dziewczynek) były dwie siostry (tab. 1).
U każdego pacjenta zbierano dokładny wywiad poszukując występowania schorzenia w rodzinie, ustalano wiek, w którym pojawiły się pierwsze zmiany skórne i objawy ze strony przewodu pokarmowego.
U wszystkich wykonano wielokrotnie badania laboratoryjne: morfologia, OB, transminazy, fosfatazę alkaliczną, poziom Fe, Cu, immunoglobulin i badanie ogólne moczu.
Wykonywano również badanie poziomu cynku w surowicy i krwinkach w laboratorium CZD metodą atomowej absorbcji w płomieniu acetylen-powietrze.
Oznaczenie poziomu Zn wykonano u 26 pacjentów minimum czterokrotnie w odstępach trzymiesięcznych. Następne badania, w tak różnorodnej grupie pacjentów, wykonywane były okazjonalnie.
Spośród 18 dorosłych pacjentów (powyżej 16 rż.) dodatkowe badanie poziomu cynku wykonano u 13 osób.
Pacjentów podzielono na dwie grupy:
– grupę pierwszą stanowiło 12 pacjentów z pełnymi objawami AE, u których pierwsze badanie poziomu cynku wykonane było przed rozpoczęciem leczenia,
– grupa druga to 14 pacjentów, u których pierwsze oznaczenie poziomu cynku wykonano w trakcie leczenia.
Leczenie rozpoczynano w różnych okresach trwania choroby. U pięciorga dzieci stosowano początkowo preparaty chinolonowe, a od 1975 roku wszyscy leczeni byli preparatami cynku (ZnSO4), podawanymi w soku owocowym. W początkowym okresie choroby u niemowląt i małych dzieci podawano 20-40 mg ZnSO4 dziennie (tj. 2,5-10 mg/kg/m.c.), dzieciom starszym 35-100 mg dziennie (tj. 3 mg/kg m.c.).
W okresie wolnym od objawów klinicznych dawkę podtrzymującą ustalono indywidualnie. Była ona niższa od leczniczej i wynosiła od 1-3 mg/kg m.c. Pacjenci dorośli wymagali leczenia okresowego w dawkach 35-90 mg/db (tj. 0,5-2 mg/kg/db), a niektórzy pozostawali bez leczenia w ciągu ostatnich kilku lat. W okresie ciąży u dwóch pacjentek konieczne było zastosowanie stałego podawania cynku w dawce 90 mg/db (tj. 1-1,5 mg/kg/db). Pozostali pacjenci wymagali zwiększenia dawki lub ponownego włączenia leczenia w czasie infekcji dróg oddechowych i dyspepsji.
Wyniki badań i omówienie
Acrodermatitis Enteropathica jest schorzeniem występującym bardzo rzadko. Dotychczas nie znaleziono w piśmiennictwie doniesień o tak długotrwałych obserwacjach chorych jak w przedstawionym materiale. AE jest chorobą uwarunkowaną genetycznie, dziedziczoną w sposób autosomalny, recesywny. Znane jest występowanie u rodzeństwa, a nie u rodziców (9, 16, 17). W naszym materiale, rodzeństwa (2 siostry i 3 braci) stanowiły 19%.
Pierwsze zmiany skórne w 77% przypadków pojawiły się w okresie niemowlęcym. Były to ogniska rumieniowo-wysiękowe z wykwitami pęcherzykowymi zajmującymi dystalne części ciała i okolice otworów naturalnych (ryc. 2, 3) oraz przedłużające się biegunki. Zwykle zbiegało się to z okresem przejścia z karmienia naturalnego na sztuczne. Niektórzy autorzy sugerują lepsze przyswajanie cynku z mleka matki i wskazują na rolę pokarmu kobiecego w leczeniu AE (3, 13, 16, 17). Często jednak schorzenie to bywa rozpoznawane późno i sprawia trudności terapeutyczne. W naszym materiale, u 30% pacjentów pierwsze objawy pojawiły się w 2-3 rż. Początkowo obserwowano u tych dzieci upośledzone łaknienie, apatię, luźne stolce, a dopiero później pojawiły się zmiany na skórze o typowej lokalizacji, pozwalające podejrzewać AE. U kilkorga z tych dzieci wystąpiło wypadanie brwi i rzęs oraz łysienie. W początkowym okresie choroby, szczególnie u niemowląt, obserwowano skłonność do zakażeń bakteryjnych, wirusowych i drożdżakowych. Ich przyczynę w części wyjaśniają wyniki badań, które wykazały, że niedobór cynku hamuje prawidłową czynność układu limfocytów T i B oraz może wywierać wpływ na chemotaksję monocytów i neutrofili (15).
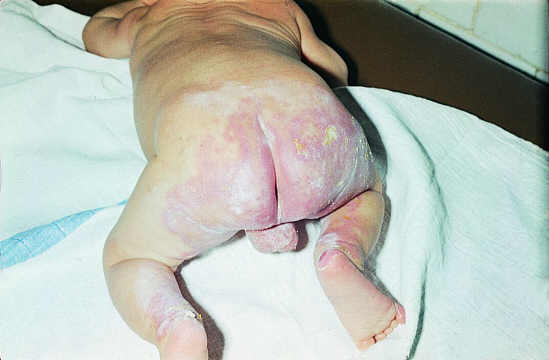
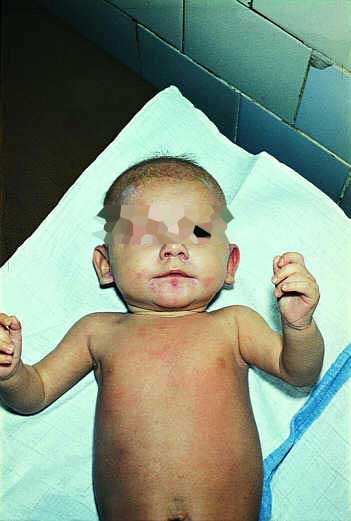
Ryc. 2, 3. Pacjent Z.A., wiek 4/12. Zmiany o charakterze rumieniowo-wysiękowym z wykwitami pęcherzykowymi wokół otworów naturalnych.
W latach sześćdziesiątych i siedemdziesiątych pięcioro pacjentów leczonych było związkami chinolinowymi. Czworo z nich dobrze znosiło wieloletnie leczenie. U jednego natomiast doszło do ciężkich, nieodwracalnych powikłań w postaci zaniku nerwu wzrokowego z utratą wzroku i niedowładu kończyn dolnych. Inni autorzy opisywali również podobne ciężkie uszkodzenia narządu wzroku i układu nerwowego (7). Według Moynahana (8, 13) stosowanie chinolonów miało przeciwdziałać wiązaniu się cynku, z „toksycznym oligopeptydem” powstającym w wyniku nieprawidłowej przemiany tryptofanu. Uwalnianie cynku miało stanowić mechanizm działania tych preparatów (8).
Od 1975 roku wszyscy opisani chorzy leczeni byli preparatami cynku. Wysokość wstępnej dawki leczniczej ustalano indywidualnie, niezależnie od wagi i wieku dziecka (ryc. 4). Początkowo wynosiła ona 2,5-10 mg/kg/db.
Wybitną poprawę obserwowano u wszystkich dzieci już po 2-3 tygodniach leczenia. Po ustąpieniu objawów chorobowych, u wszystkich dzieci zmniejszano dawkę do podtrzymującej, która kontrolowała zmiany skórne i objawy ze strony przewodu pokarmowego.
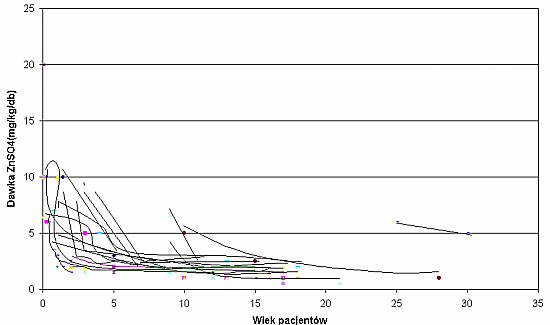
Ryc. 4. Wyniki wieloletnich obserwacji wysokości stosowanych dawek ZnSO4 kontrolujących zmiany skórne u pacjentów z AE.
Na przestrzeni wieloletnich obserwacji zauważono, że należy indywidualnie ustalać dzienną dawkę ZnSO4, kontrolującą objawy chorobowe, niezależnie od wagi i wieku. Jej wysokość u każdego dziecka okazała się różna i wynosiła około 1-3 mg/kg m.c. Dawkę tę stosowano skutecznie przez wiele miesięcy i lat, natomiast w czasie dyspepsji i infekcji konieczne było okresowe jej zwiększanie.
Wyniki tych obserwacji pozwoliły na stosowanie u naszych pacjentów preparatów cynku w dawkach znacznie niższych od zalecanych przez innych autorów (8). Uważali oni, że najskuteczniejszą dawką ZnSO4, którą należy stosować przez wiele miesięcy jest 5 mg/kg/m.c. (8).
Należy podkreślić, że wszystkie obserwowane dzieci, znosiły dobrze leczenie cynkiem i nie obserwowano żadnych objawów ubocznych.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Abitan R. et al.: Carrence acquise en zinc chez un nourrson premature nourri an sein. Ann. Dermatol. Venerol. 1994, 121:635-638. 2. Danbolt C.J., Closs K.: Acrodermatitis Enterophatica, Acte Derm. Venerol (Stockh.) 1942, 23:127. 3. Evans G.W., Johnson P.E.: Zinc-binding factor in Acrodermatitis Enterophatica Lancet 1976, 2:1310. 4. George P. et al.: Acroderm. Entero. – like syndrom secondary to Isoleucine Deficiency during treatment of maple simp. Urine Disease. AJDC 1993, 147, 954, 956. 5. Ghali Fred et al.: Picture of the Month. Arch. Pediatrics Adolescens Med. 1996, 150, 99, 100. 6. Jabłońska S.: Choroby skóry. PZWL Warszawa 1980, 663. 7. Michaelson C.: Zinc therapy in AE, Acta Derm. Venerol. (Stockh.) 1974, 54: 377-382. 8. Moynahan E.J.: Acrodermatitis Enteropathica lethal inherited human Zinc – deficiency discorder. Lancet 1974, 2:399. 9. Piórkowska E.: Przypadek Acrodermatitis Enteropathica u niemowlęcia. Wiadomości Lekarskie 1988, 41, 5:320. 10. Piela Z. i wsp.: Nabyty niedobór cynku u wcześniaka karmionego wyłącznie mlekiem matki. Post. Dermatol. 1997, 14: 415-412. 11. Rosińska-Borkowska D., Chrupek-Gozdek A.: Acrodermatitis Enteropathica w świetle własnych obserwacji. Przegl. Dermatol. 1990, 77:6. 12. Rosińska-Borkowska D., Chrupek-Gozdek A.: Wyniki wieloletnich obserwacji Acrodermatitis Enteropathica u 18 pacjentów. Ped. Pol. 1994, 99:104. 13. Rubisz-Brzezińska J. i wsp.: Acrodermatitis Enteropathica – zespół niedoboru cynku. Przegl. Dermatol. 1977, 64:465-469. 14. Sharma N.L. et al.: Self limting Acrodermatitis Enteropathica. Int. J. Dermatol. 1988, 277-14. 15. Weston S.L. et al.: Zinc correction of detective chemotaxis in Acrodermatitis Enteropathica. Arch. Dermatol. 1977, 113:422, 426. 16. Zimmerman A. et al.: Acrodermatitis in breast-fed premature infants: evidence for a defect of mammary Zinc secretion. Pediatrics 1982, 69:2-16. 17. Zychowicz Cz., Siewierska B.: Wartość pokarmu kobiecego w leczeniu Acrodermatitis Enteropathica w świetle teorii niedoboru cynku. Ped. Pol. 1978, 23:315-321.