© Borgis - Nowa Pediatria 3/2002, s. 174-178
Anna Buchner1, Franciszek Iwańczak2
Etiopatogeneza wrzodziejącego zapalenia jelita grubego
Etiopathogenesis of ulcerative colitis
1z Christ Medical Center, Dak Lawn, Illinois
2z II Katedry i Kliniki Pediatrii, Gastroenterologii i Żywienia Akademii Medycznej we Wrocławiu
Kierownik Kliniki: prof. dr hab. n. med. Franciszek Iwańczak
Streszczenie
In the present review the genetic, infections, dietetic and environmental factors contributing to etiopathogenesis of ulcerative colitis are discussed. The role of cytokines in pathogenesis of the disease is reviewed more extensively.

Wrzodziejące zapalenie jelita grubego (wzjg) jest przewlekłym nieswoistym procesem zapalnym błony śluzowej jelita grubego o niewyjaśnionej etiologii i najczęściej przewlekłym przebiegu, z okresami zaostrzeń i remisji. Proces zapalny obejmuje jelito grube, zawsze dotyczy odbytnicy, może rozszerzać się na okrężnicę, nie zajmując jelita cienkiego. Wrzodziejące zapalenie jelita grubego jest chorobą ludzi młodych. Najwięcej zachorowań występuje pomiędzy 20 a 40 rokiem życia, około 15-20% przypadków rozpoczyna się u dzieci i młodzieży szkolnej. Na podstawie licznych badań przyjmuje się, że częstość występowania wzjg w ostatnich latach jest stabilna i wynosi 1,5-10,0/105 ludności. Częstość występowania i zapadalność na wzjg różni się w zależności od rejonu geograficznego i rasy. Nieswoiste zapalenia jelit (wrzodziejące zapalenie jelita grubego, choroba Leśniowskiego-Crohna) występują najczęściej u mieszkańców Skandynawii, Europy Zachodniej i Ameryki Północnej, znacznie rzadziej u ludności Azji, Afryki i Ameryki Południowej. Zdecydowanie częściej stwierdza się chorobę w populacji rasy białej oraz Żydów w porównaniu z innymi grupami etnicznymi, żyjącymi w tych samych warunkach (1, 2). W większości publikacji wskazuje się na jednakową częstość występowania u kobiet i mężczyzn (3).
ETIOLOGIA WRZODZIEJĄCEGO ZAPALENIA JELITA GRUBEGO
Etiologia wrzodziejącego zapalenia jelita grubego jest nieznana. Choroba jest najprawdopodobniej uwarunkowana przez wiele czynników i różnorodność mechanizmów mogących mieć udział w jej wywołaniu. Wśród tych czynników wymienia się: czynniki genetyczne, infekcje, środowiskowe, dietetyczne, procesy immunologiczne, zaburzenia metabolizmu krótkołańcuchowych kwasów tłuszczowych w kolonocytach, działanie wolnych rodników, tlenku azotu, zmiany w składzie śluzu i inne (4-7). Mechanizmy etiopatogenetyczne wrzodziejącego zapalenia jelita grubego zostały przedstawione na rycinach 1, 2, 3.
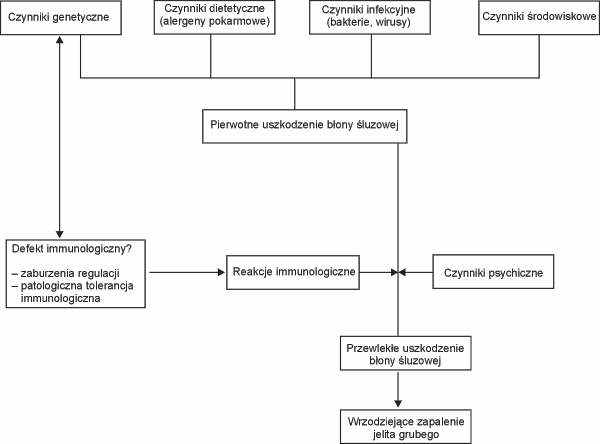
Ryc. 1. Etiopatogeneza wrzodziejącego zapalenia jelita grubego.
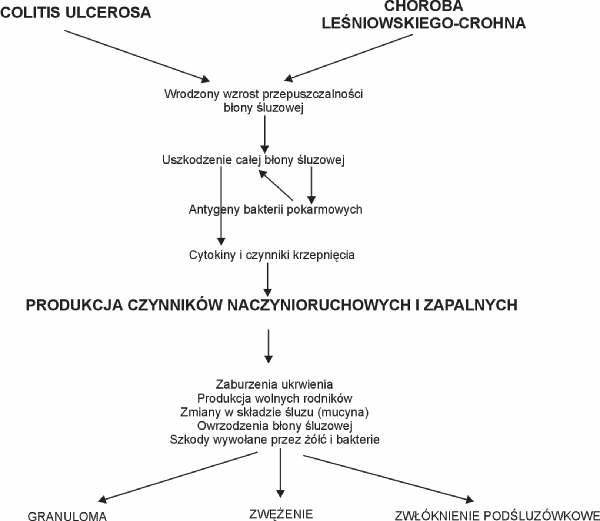
Ryc. 2. Mechanizmy powstawania zmian patologicznych w nieswoistych zapaleniach jelit.
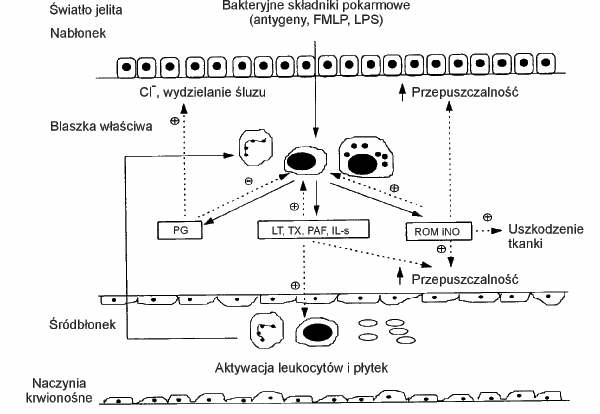
Ryc. 3. Schemat przedstawiający działanie antygenów bakteryjnych i pokarmowych na wyzwalanie zapalenia błony śluzowej jelita (LPS - lipopolisacharydy; FMLP - zwiazek chemiczny; PG - prostaglandyny; LT - leukotrieny; TX - tromboksan; PAF - czynnik aktywujący płytki; IL-s-interleukiny; ROM - metabolity działania tlenu, iNO - indukowana syntaza tlenku azotu).
Za udziałem czynników genetycznych przemawiają różnice rasowo-etniczne w częstości występowania tej choroby. Co więcej, różna zapadalność na chorobę w różnych grupach etnicznych i związek z zespołami i chorobami genetycznymi, ze znanymi predyspozycjami wskazuje, że istnieje predyspozycja genetyczna w rozwoju nieswoistego zapalenia jelit (8, 9). Rasa biała choruje 2-4 razy częściej niż ciemnoskórzy. Obserwuje się zwiększoną zachorowalność wśród członków rodzin pacjentów, przede wszystkim wśród krewnych pierwszego stopnia. W tej grupie zachorowalność jest wyższa o 10-30% niż w populacji ogólnej, przy niezwiększonej zachorowalności wśród współmałżonków, co może pośrednio przemawiać przeciwko udziałowi czynników infekcyjnych. Opisano częstsze występowanie choroby u bliźniąt jednojajowych (10). U chorych na wrzodziejące zapalenie jelita grubego opisano częstsze występowanie allelu DR2 HLA II oraz związek występowania tych alleli z obecnością przeciwciał przeciw cytoplazmie granulocytów obojętnochłonnych (ANCA) (11).
Stokkres i wsp. (12) poszukiwali możliwych związków pomiędzy fenotypem HLA klasy II a nieswoistymi zapaleniami jelit. Na podstawie przeglądu piśmiennictwa stwierdzili, że wrzodziejące zapalenie jelita grubego wykazuje dodatnią korelację z HLA-DR2, DR9 oraz-DRB10 103. Satsangi i wsp. (13) również wykazali korelację między predyspozycją do rozwoju choroby a występowaniem genomu HLA DR2. Inne wyniki badań wskazują na związek między pewnymi regionami chromosomów 2. i 6. a skłonnością do występowania wzjg, jak również między regionami położonymi na chromosomie 3, 7 i 12 a predyspozycją do rozwoju wzjg i ch. L-C (14). W badaniach epidemiologicznych wykazano, że przebyte w okresie okołoporodowym lub w pierwszych latach życia zakażenie wirusami odry, świnki i innymi są czynnikami ryzyka rozwoju nieswoistych zapaleń jelit w późniejszym okresie życia (15-17). Ważną rolę może także odgrywać modulowana immunologicznie aktywacja genów cytokin (18,19), która u pacjentów z chorobą Leśniowskiego-Crohna stymuluje komórki jednojądrzaste do produkcji cytokin i ekspresji ich receptorów (20). Sugeruje to, że geny cytokin odgrywają istotną rolę w przebiegu nieswoistych zapaleń jelit.
Również swoiste i nieswoiste czynniki środowiskowe i odżywcze mogą powodować wystąpienie lub zaostrzenie chorób zapalnych jelit. Ostatnio wykazano, że częstość występowania choroby Leśniowskiego-Crohna w dużych miastach jest większa niż na wsi. Sugeruje się, że pacjenci z chorobą L-C spożywają więcej sacharozy, węglowodanów prostych oraz omega-6 kwasów tłuszczowych, natomiast mniej owoców i warzyw. Również w diecie miejskiej znajdują się duże ilości nieaktywnych, nieorganicznych mikrocząsteczek, konserwanty oraz środki spulchniające, które z lipopolisacharydami ścian bakteryjnych tworzą cząsteczki o właściwościach antygenowych, mogące modulować miejscową i układową odpowiedź immunologiczną. Zastosowanie diety pozbawionej mikrocząsteczek powodowało znaczące zmniejszenie aktywności choroby (20).
Według jednej z hipotez przyczyną wzjg są zaburzenia metabolizmu krótkołańcuchowych kwasów tłuszczowych w kolonocytach, prowadzące do upośledzenia wytwarzania śluzu, wzrostu przepuszczalności błony śluzowej jelita, przedłużania się ekspozycji na antygeny środowiskowe, aktywację śluzówkowego układu immunologicznego i przewlekania się procesu zapalnego (21, 22).
Teoria infekcyjna przypisuje dużą rolę florze bakteryjnej przewodu pokarmowego. Wymienia się bakterie E. coli, która produkuje enzymy degradujące mucynę oraz H2S wpływający na metabolizm krótkołańcuchowych kwasów tłuszczowych, zakażenie Clostridium difficile i niektóre gatunki Mycobacterium. Wyniki badań przeprowadzone na modelach zwierzęcych pozwalają przypuszczać, że do zapoczątkowania procesu zapalnego i jego utrzymywania się niezbędna jest obecność flory jelitowej. W hodowli myszy pozbawionych interleukiny-10 lub Il-2, prowadzonych w środowisku pozbawionym drobnoustrojów nie występowały kliniczne objawy zapalenia jelit. Po wprowadzeniu normalnej flory bakteryjnej rozwijało się zapalenie jelit (3).
W patogenezie nieswoistych zapaleń zwraca się również uwagę na rolę przeciwciał bakteryjnych, które reagują krzyżowo z tkanką jelita, rolę granulocytów obojętnochłonnych czy tlenku azotu (23, 24, 25, 26).
Istnieją również dowody potwierdzające rolę jelitowego układu nerwowego w patogenezie nieswoistego zapalenia jelit. Stres może zaostrzać zapalenie powodując ważne interakcje pomiędzy ośrodkowym układem nerwowym, jelitowym układem nerwowym i przewodem pokarmowym. Zarówno w chorobie Leśniowskiego-Crohna jak i we wrzodziejącym zapaleniu jelita grubego wzrasta immunoreaktywność substancji P. Jak wynika z badań doświadczalnych substancja P w jelicie prawdopodobnie działa prozapalnie a antagoniści receptorów P zmniejszają naciek granulocytarny w jelicie zapalnym szczura spowodowanym kwasem trójnitrobenzosulfanylowym (27, 28). Podanie miejscowe i podskórne lignokainy wyraźnie zmniejsza odczyn zapalny, a w niesprawdzonym placebo badaniu wlewy doodbytnicze z lignokainy skutecznie leczyły pacjentów z owrzodzeniami jelita grubego (29). Uważa się, że lignokaina hamując przewodzenie nerwowe może blokować bodźce nerwowe przewodzone przez jelitowy układ nerwowy (6, 30).
Najwięcej zwolenników posiada jednak pogląd, że główną rolę w patogenezie nieswoistego zapalenia jelit pełnią procesy immunologiczne, w których zaangażowane są mechanizmy odpowiedzi komórkowej i humoralnej. W wzjg dochodzi do zaburzenia w populacji limfocytów T, wzmożonej aktywności miejscowego systemu immunologicznego i zwiększonego wytwarzania prozapalnych cytokin (31, 32).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Andres P.G., Friedman L.S.: Epidemiology and the natural course of inflammatory bowel disease. Gastroenterol. Clinic North Am. 1999, 25:255-281. 2. Satsangi J. et al.: Genetics of imflammatory bowel disease. Gut 1994, 35, 696-700. 3. Moses P.L. i wsp.: Choroby zapalne jelit. Przyczyny, objawy, przebieg. Medycyna po Dyplomie, 1999, 2-6. 4. von Allmen D. et al.: Inflammatory bowel disease in choldren. Curr. Opin. Pediatr. 1995, 7:547-552. 5. Fiocchi C.: Inflammatory bowel disease: Etiology and pathogenesis. Gastroenterology, 1998, 115:182-205. 6. Goyal R.K., Hirano I.: The enteric nervous system. N. Engl. J. Med. 1996, 334:1106-1115. 7. Integlia M.J. et al.: Pediatric inflammatory bowel disease. Cur. Opin. Gastroenterol. 1996, 12:345:351. 8. Matsuura T. et al.: Immune activation genes in inflammatory bowel disease. Gastroenterology 1993, 104:448-458. 9. Pena A.S. et al.: Genetics and epidemiology may contribute to understanding the pathogenesis of IBD – a new approach is now indicated. Can J. Gastroenterol. 1993, 7:71-75. 10. Koutroubakis J. et al.: Immunogenetics of cytokines. Relevance for future research on inflammatory bowel disease. Scand. J. Gastroenterol. 1995, 30:1139-1146. 11. Rump J.A. et al.: Ein neues ANCA – Muster in Seren Patienten mit Colitis ulcerosa. Immun. Infekt. 1992, 20:16-19. 12. Stokkers P.C.F. et al.: HLA-DR and DQ phenotypes in inflammatory bowel disease: a meta-analysis. Gut 1999, 45:395-401. 13. Satsangi J. et al.: Two stage genome-wide search in inflammatory bowel disease: evidence for susceptibility loci on chromosomes 3,7 and 12, Nature Genet. 1996, 14:199-202. 14. Satsangi J. et al.: Contibution of genes of the major histocompatibilty complex to susceptibility and disease phenotype in inflammatory bowel disease. Lancet 1996, 347:1212-1217. 15. Ekbom A. et al.: Perinatal measles infection and subsequent Crohn?s disease. Lancet 1994, 344:508-510. 16. Fiocchi C.: Imflammatory bowel disease: etiology and pathogenesis. Gastroenterology 1998, 115:182-205. 17. Montgomery S.M. et al.: Paramyxovirus infections in childhood and subsequent inflammatory bowel disease. Gastroenterology 1999, 116:776-803. 18. Neurath M.F. et al.: Antibody to I1-2 abrogates established experimental colitis in mice. J. Exp. Med. 1995, 182:1281-1290. 19. Stuber E. et al.: Blocking the CD 40L-CD40 interaction in vivo specifically prevents the priming of Th-T cells through the inhibition of I1-12 secretion. J. Exp.Med. 1996, 183:183-189. 20. Matsuura T. et al.: Immune activation genes in inflammatory bowel disease, Gastroenterology 1993, 104, 448-458. 21. Fiocchi C.: Cytokines. In: Targan S.R., Shanahan F., editors. Inflammatory bowel disease. From bench to bedside. Baltimore: Williams & Wilkins, 1994, 106-122. 22. Gross V. et al.: Inflammatory mediators in chronic inflammatory bowel diseases. Klin. Wschr. 1991, 69:981-987. 23. Berberian L.S. et al.: Expression of a novel autoantibody defined by the VH3-15 gene in inflammatory bowel disease and Campylobacter jejuni enterocolitis. J. Immunol. 1994, 153:3756-3763. 24. Carter L., Wallace J.L.: Alteration in rat peripheral blood neutrophil function as a consequence of colitis. Dig. Dis. Sci. 1995, 40, 192. 25. Hoppen Th. et al.: Clostridium difficile bei frühkindlicher Pancolitis ulcerosa. Monatsschr. Kinderheilkd. 1993. 141:474-477. 26. Marcinkiewicz J.: Regulation of cytokine Production by eicosanoids and Nitric Oxide. Arch. Immunol. Ther. Exp. 1997, 45:163-167. 27. Harada K. et al.: Role of cytokine induced neutrophilic chemoatractant, a member of the interleukin-8 family, in rat experimental colitis. Digestion. 1994, 55, 179. 28. Morris G. et al.: Hapten induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology 1989, 96:795. 29. Veljaca M. et al.: BPC-15 reduces trinitrobenzene sulphonic acid induced colonic damage in rats. J. Pharmacol. Exp. Ther. 1995, 272, 417. 30. Gaboes K., Collins S.: Structural abnormalities nervous system in Crohn´s disease and ulcerative colitis. Neurogastroenterol. Mot. 1998, 10:189-202. 31. Birkedal-Hansen H.: Role of cytokines and inflammatory mediators in tissue destruction. J. Periodont. Res. 1993, 28:500 -510. 32. Kam L. et al.: Cytokines and chemokines in inflammatory bowel disease. Cur. Sci. 1995, 11:305:309. 33. Radford-Smith G., Jewell D.P.: The role of cytokines in inflammatory bowel disease. Mediators Inflam 1994, 3:3-9. 34. Sartor R.B.: Cytokines in intestinal inflammation: pathophysiological and clinical considerations. Gastroenterology 1994, 106:533-539. 35. Fell J.M.E., Walker-Smith J.A., Spencer J., Macdonald T.T.: The distrbution of dividing T cells throughout the intestinal wall in inflammatory bowel disease (IBD). Clin Exp. Immunol. 1996, 104:280-285.