© Borgis - Postępy Fitoterapii 2/2007, s. 95-108
Aleksandra Chrobot, *Adam Matkowski
Roślinne produkty naturalne jako blokery kanałów wapniowych układu sercowo-naczyniowego
Plant natural drugs as calcium channel blockers in the cardiovascular system
Katedra Biologii i Botaniki Farmaceutycznej Akademii Medycznej we Wrocławiu
Kierownik Katedry: dr farm. Alicja Noculak-Palczewska
Summary
Calcium channel blockers are an important group of cardiovascular drugs. They inhibit the calcium influx into the smooth muscle cells and the cardiomyocytes, thus decreasing the contractibility. They act as negative chronotropic and inotropic agents. The physiological effects are: reduced blood pressure, improvement of coronary and peripheral circulation, as well as antiarrythmic activity.
In the recent years, numerous studies have confirmed that many ethnomedicinal plant natural products used in cardiovascular diseases, exhibit the ability to inhibit the calcium channels in vascular and heart muscle cells. Additionally, these compounds can modulate the intracellular calcium by inhibition of the release from ER. As the result, they cause the analogous pharmacological effects as the established synthetic Ca-blockers.
Besides, most plant derived drugs have multidirectional influence on human physiology, which extends their activity spectrum.
Calcium channel blocker of plant origin are from the chemical structure point of view very variable.
Following groups can be distinguished:
1. Alkaloids:
– aporphine - nantenine i liriodenine,
– bis-benzyl-isoquinoline - tetrandrine, fangquinoline, dauricine, daurisoline, nepherine i hernandesine,
– indole - hirsutine and Himalanthus lancifolius (Apocynaceae) alkaloids
– pyrazine - ligustrazine.
2. Triterpene saponins: ginsenosides.
3. Isoflavones: genistein, daidzein, equol i puerarin.
4. Coumarins and furanochromon derivatives: khelline, visnagin and visnadin.
5. Terpenoids: ginkgolides, marrubenol i S-petasine.
6. Miscellaneous: resveratrol, hinokiol, magnolol and butylidenephthalide.
Several studies indicate the calcium antagonist activity in other plants such as: Alstonia scholaris (Apocynaceae), Salvia miltiorrhiza (Lamiaceae) i Dalbergia odorifera (Leguminosae). However, the information of chemical structures responsible for that activity is still not available.
The calcium blockers of plant origin have great potential as cardiovascular drugs but only few are sufficiently documented clinically. Most of them have been investigated in vitro or in animal studies. In the near future, the importance and consumption of the plant complementary medicines would most likely increase, as soon as more consistent clinical data become available.

Blokery kanałów wapniowych są ważną grupą leków stosowanych w schorzeniach układu sercowo-naczyniowego. Hamując napływ jonów wapnia do komórek mięśni gładkich i mięśnia sercowego, zmniejszają ich kurczliwość, co skutkuje obniżeniem ciśnienia krwi, działaniem chrono- i inotropowym, poprawą krążenia wieńcowego i obwodowego oraz działaniem antyarytmicznym.
W ostatnich latach stwierdzono, że wiele związków, wyizolowanych z roślin znajdujących zastosowanie w medycynie ludowej do leczenia schorzeń układu sercowo-naczyniowego, wykazuje zdolność blokowania napływu jonów wapnia do komórki przez blokowanie kanałów wapniowych. Wpływają one też na stężenie wapnia wewnątrzkomórkowego przez hamowanie jego uwalniania z magazynów siateczki śródplazmatycznej. Efektem tego działania jest aktywność farmakologiczna, analogiczna do aktywności syntetycznych blokerów kanałów wapniowych. Związki pochodzenia roślinnego charakteryzują się wielokierunkowym wpływem na organizm ludzki, co rozszerza ich spektrum działania.
Blokery kanałów wapniowych
Zmiany w ilości jonów wapnia i ich transporcie przez błony są często powiązane ze stanami patologicznymi, włączając schorzenia układu sercowo-naczyniowego (1, 2). Związki blokujące kanały wapniowe, zwane blokerami kanałów wapniowych, odgrywają istotną rolę w leczeniu schorzeń układu sercowo-naczyniowego. Komórkowe szlaki transdukcji sygnałów, w których pośrednikiem są jony wapnia, stały się zatem ważnym celem poszukiwań skutecznych terapeutyków (3). Obecnie stosowane syntetyczne związki blokujące kanały wapniowe wykazują działanie głównie w stosunku do kanałów wapniowych typu L, jednakże w większych stężeniach mogą działać również na pozostałe typy kanałów wapniowych.
Mechanizm działania blokerów kanałów wapniowych polega na allosterycznej stabilizacji kanału w stanie nieaktywnym. Miejsce wiążące blokery kanałów wapniowych stanowi podjednostka α1. Związki, w zależności od budowy chemicznej, mogą związać się z nią w jednym z trzech miejsc wiążących. Połączenie się leku z podjednostką α1 powoduje zmianę jej konformacji i zablokowanie kanału dla jonów wapnia (1). Poszczególne grupy antagonistów kanałów wapniowych mają odrębne miejsca wiążące w obrębie podjednostki α1. Mogą one być zlokalizowane na zewnątrz komórki, czyli związek ma możliwość połączenia się niezależnie od częstotliwości otwierania kanału wapniowego. Mogą także znajdować się w świetle kanału, więc są dostępne tylko po wniknięciu cząsteczki leku przez por kanału. Oznacza to, że związki te są aktywne tylko po pobudzeniu kanału wapniowego, a ich siła działania wzrasta wraz z częstotliwością otwierania kanału (4).
Budowa chemiczna determinuje także różnice w powinowactwie do miejsc, w których występują kanały wapniowe. Zatem blokery kanałów wapniowych mogą wykazywać większą aktywność farmakologiczną w stosunku do mięśni gładkich naczyń krwionośnych lub mięśnia sercowego. Efektem działania blokerów jest rozkurcz mięśni gładkich w naczyniach. Związane z tym obniżenie ciśnienia krwi oraz zmniejszenie siły skurczu mięśnia sercowego, zmniejsza zapotrzebowanie na tlen. Innym efektem jest działanie przeciwarytmiczne (1).
Przegląd związków pochodzenia naturalnego o aktywności blokerów kanałów wapniowych
Ostatnie dekady zaowocowały opracowaniem licznej grupy związków syntetycznych o aktywności antagonistów jonów wapnia jako nowych leków skutecznych w terapii schorzeń układu sercowo-naczyniowego. Ich mechanizm działania wzbudził zainteresowanie poszukiwaniami naturalnych antagonistów jonów wapnia. Stwierdzono, że wiele znanych i od dawna stosowanych leków roślinnych wykazuje taką aktywność (3). Biomedyczne i technologiczne osiągnięcia ostatnich lat umożliwiły przebadanie, identyfikację i scharakteryzowanie poszczególnych związków oraz poznanie mechanizmu, leżącego u podstaw właściwości terapeutycznych, powiązanych z tymi środkami od wieków (5). Nie wszystkie przypuszczenia o działaniu antagonistycznym w stosunku do jonów wapnia, wykazywanym przez leki tradycyjne, zostały potwierdzone. W zasadzie niewiele z nich ma lepsze terapeutyczne lub ekonomiczne wartości w porównaniu z obecnie stosowanymi lekami syntetycznymi. Niemniej, badania nad aktywnością składników roślin leczniczych dostarczały wielu nowych obserwacji i interesujących odkryć (3).
Wpływ większości produktów naturalnych na schorzenia układu sercowo-naczyniowego jest, w odróżnieniu od leków syntetycznych, wieloraki i złożony. W modelach zwierzęcych wykazują one zdolność rozkurczania mięśni gładkich naczyń krwionośnych, hamowania kurczliwości mięśnia sercowego i poprawy jego funkcji oraz wyciszania arytmii, wywołanej przez niedokrwienie, a nawet zmniejszania obszaru zawału mięśnia sercowego. Dodatkowo, niektóre z nich wykazują unikalny profil antyoksydacyjny i inne, pozytywne efekty działania, co wzmacnia ich pozycję w terapii schorzeń serca i naczyń krwionośnych (6).
Zaprezentowany tu przegląd związków pochodzenia naturalnego o aktywności antagonistów jonów wapnia nie wyczerpuje w całości zagadnienia, jednakże przedstawia najważniejsze i najlepiej zbadane składniki o takim działaniu.
Alkaloidy pochodne aporfiny
Pochodne aporfiny są grupą alkaloidów izochinolinowych, których główny szkielet składa się z ugrupowania izochinolinowego i układu fenantrenu (ryc. 1). Występują w rodzinach: Annonaceae, Berberidaceae, Lauraceae, Magnoliaceae, Menispermaceae, Monimiaceae, Nymphaeaceae, Papaveraceae, Ranunculaceae oraz Rutaceae (7).
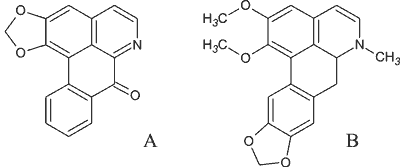
Ryc. 1. Alkaloidy aporfinowe: A – liriodenina, B – nantenina.
Obecna w kilku gatunkach roślin (+)-nantenina wykazuje właściwości antagonistyczne w stosunku do receptorów α1-adrenergicznych. (+)-Nantenina, wyizolowana z Platycapnos spicata (Papaveraceae), proporcjonalnie do stężenia, hamuje czynność skurczową, indukowaną noradrenaliną lub wysokim stężeniem chlorku potasu w aortach szczurów (8). Efekty farmakologiczne (+)-nanteniny, obserwowane przy stężeniu niższym niż 1 μM, są powiązane z działaniem antagonistycznym na receptory α1-adrenergiczne i receptory serotoninowe 5-HT2A. Natomiast u podstaw aktywności farmakologicznej (+)-nanteniny w stężeniu powyżej 1 μM leży hamowanie napływu jonów wapnia przez kanały wapniowe, inhibicja PKC (kinazy białkowej C), a także blokowanie receptorów α2-adrenergicznych (8).
Działanie antagonistyczne w stosunku do jonów wapnia w komórkach mięśni gładkich aorty szczura wyjaśnia, przynajmniej częściowo, efekt chronotropowo ujemny, wykazywany przez (+)-nanteninę w stężeniach powyżej 1 μM w wyizolowanym przedsionku serca szczura. Biorąc pod uwagę właściwości farmakologiczne (+)-nanteniny, wykazane in vitro i obserwowane in vivo, istnieje możliwość stosowania tego związku jako leku hipotensyjnego (8).
Alkaloidem aporfinowym jest również liriodenina, wyizolowana z Fissistigma glaucescens (Annonaceae). Oprócz działania antagonistycznego w stosunku do receptorów M3 w mięśniach gładkich, wykazuje także potencjał antyarytmiczny i działanie inotropowo dodatnie. Jest ona nieselektywnym blokerem kanałów jonowych. Działa hamująco zarówno na prąd sodowy, potasowy, jak i na prąd wapniowy typu L, przy czym najsłabiej hamuje ten ostatni. W wyizolowanych mięśniach komory i przedsionka serca liriodenina zwiększa siłę skurczu i spowalnia spontaniczny rytm prawego przedsionka (9).
Alkaloidy bis-benzylizochinolinowe
Alkaloidy bis-benzylizochinolinowe należą do grupy alkaloidów izochinolinowych o charakterze dimerycznym. Mają dwa rdzenie benzylizochinolinowe połączone mostkami eterowymi (ryc. 2). Liczba łączących mostków jest różna i może wahać się od jednego do trzech. Związki te występują najczęściej w roślinach z rodzin: Menispermaceae, Berberidaceae, Magnoliaceae, Monimiaceae, Annonaceae i Combretaceae (7).
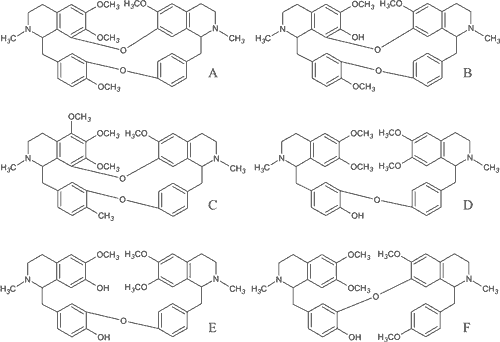
Ryc. 2. Alkaloidy bis-benzylizochinolinowe: A – tetrandryna, B – fangchinolina, C – hernandezyna, D – daurycyna, E – dauryzolina, F – neferyna.
W ciągu kilku ostatnich dekad wykonano dużą liczbę badań, mających na celu określenie aktywności farmakologicznej tej grupy związków, od dawna stosowanych w medycynie ludowej, zwłaszcza w Chinach. Szczególnie duże znaczenie wykazują alkaloidy otrzymane ze Stephania tetrandra.
Tetrandryna. Jednym z najlepiej scharakteryzowanych alkaloidów bis-benzylizochinolinowych jest (+)-tetrandryna, wyizolowana z rośliny Stephania tetrandra z rodziny Menispermaceae. Zbudowana jest z dwóch jednostek benzylizochinoloinowych połączonych dwoma mostkami eterowymi i jest konformerem α-β (5).
Tetrandryna jest głównym składnikiem wyciągu z korzenia Stephania tetrandra, który był od wieków stosowany w medycynie chińskiej jako środek przeciwnadciśnieniowy, przeciwzapalny, przeciwgorączkowy i przeciwbólowy. Pierwsze badania farmakologiczne i toksykologiczne tetrandryny zostały opublikowane w 1937 r. i stwierdzały jej właściwości hipotensyjne i kardiodepresyjne (10).
Dalsze badania nad aktywnością farmakologiczną tetrandryny wykazały, że oprócz działania hipotensyjnego, będącego skutkiem rozkurczenia mięśniówki naczyń (10, 11) oraz działania chronotropowo i inotropowo ujemnego, wykazuje ona aktywność antyarytmiczną (5, 12). U podstawy tych działań leży zdolność tetrandryny do blokowania kanałów wapniowych typu L i typu T (3, 5, 10, 11, 12) oraz hamowania uwalniania jonów wapnia zmagazynowanych w siateczce endoplazmatycznej (3, 11).
Tetrandryna ma zdolność obniżania ciśnienia bez wpływu na rytm serca, co zostało potwierdzone u znacznej populacji pacjentów z nadciśnieniem (3). Wynika to z mniejszej efektywności tetrandryny jako blokera kanałów wapniowych w przedsionku serca niż w aorcie i sugeruje, że tetrandryna stosowana w dawce terapeutycznej w nadciśnieniu nie wpływa na zmianę rytmu serca (13).
Jednakże eksperymenty in vitro i in vivo ujawniły, że tetrandryna, stosowana w większych dawkach, ma zdolność spowalniania rytmu serca, czego skutkiem jest długotrwałe działanie ochraniające mięsień sercowy, poprzez obniżenie zapotrzebowania na tlen. Dzięki temu jest ona skuteczna w łagodzeniu choroby wieńcowej serca (10). Ponadto tetrandryna zmniejsza obszar niedokrwienia i obumarcia oraz pobudza funkcję serca po zawale mięśnia sercowego (5, 12).
Badania wpływu tetrandryny na kurczliwość naczyń krwionośnych wykazały pewną dwoistość. Mianowicie, działając bezpośrednio na mięśnie gładkie naczyń krwionośnych, czyli blokując obecne w nich kanały wapniowe typu L, powoduje rozkurcz mięśni naczyń. Natomiast przez blokowanie kanałów potasowych aktywowanych wapniem w śródbłonku naczyń, wywołuje depolaryzację błon komórkowych i skurcz mięśniówki naczyń krwionośnych (10).
Aktywność tetrandryny, jako naturalnego blokera kanałów wapniowych, wynika z wielopunktowego oddziaływania z kanałem wapniowym typu L. Tetrandryna wpływa na przyłączanie się ligandu do wszystkich trzech miejsc kompleksu receptorowego dla blokera kanału. Łącząc się bezpośrednio z podjednostką α1, zwiększa powinowactwo i siłę związania dihydropirydyny z miejscem wiążącym (5).
Tetrandryna wykazuje także zdolność oddziaływania z miejscem wiążącym pochodne benzotiazepiny, takie jak diltiazem, mimo że ma odmienną od nich budowę chemiczną (5, 14). W oparciu o profil farmakologiczny tetrandryna została sklasyfikowana jako antagonista kanałów wapniowych o właściwościach podobnych do werapamilu (14).
Istnieją doniesienia, że działanie ochronne tetrandryny na serce i układ krążenia nie jest jedynie wynikiem blokowania kanałów wapniowych, ale jest tu także zaangażowany mechanizm stymulacji receptora muskarynowego (12). Wyniki niektórych badań wskazują także, że tetrandryna oddziałuje z α1- i α2-adrenoreceptorami, blokując te struktury (3). Jednakże głównym mechanizmem działania tetrandryny pozostaje antagonizm w stosunku do kanałów wapniowych.
Drugim ważnym składnikiem korzenia Stephania tetrandra jest fangchinolina, od dawna stosowana w Chinach w terapii nadciśnienia. Mimo, że wykazano jej zdolność do hamowania napływu jonów wapnia do komórki, jest ona bardzo słabym antagonistą kanałów wapniowych (15).
W fitoterapii często stwierdza się korzystniejsze działanie pełnych wyciągów roślinnych lub ich mieszanek w stosunku do pojedynczych związków wchodzących w ich skład. Potwierdza to porównanie aktywności farmakologicznej wyciągu z korzenia rośliny Stephania tetrandra z działaniem jej poszczególnych składników, tetrandryny i fangchinoliny.
Wyciąg z korzenia Stephania tetrandra działa jak bloker kanałów wapniowych, obniżając ciśnienie tętnicze krwi, kurczliwość mięśnia sercowego i polepszając przepływ wieńcowy, a także wyrównując arytmię i zmniejszając obszar zawału mięśnia sercowego. Działanie ekstraktu ma podobne natężenie jak działanie samej tetrandryny, mimo że jej zawartość w wyciągu sięga tylko około 7% (16). Powyższe obserwacje wskazują, że tetrandryna nie jest jedynym komponentem odpowiedzialnym za działanie wyciągu z korzenia Stephania tetrandra i mamy do czynienia z działaniem synergistycznym.
Stwierdzono też, że wyciąg z korzenia Stephania tetrandra znacznie lepiej hamuje uwalnianie białek podczas tzw. paradoksu wapniowego, w porównaniu z czystą tetrandryną. Wytłumaczeniem tego zjawiska jest odkrycie, że mieszanina tetrandryny i fangchinoliny w stosunku 50:50 wykazuje prawie identyczny efekt jak sama tetrandryna. Sugeruje to istnienie interakcji synergistycznej między dwoma składnikami ekstraktu (15).
Odkryto też dodatkowy mechanizm antagonizmu w stosunku do jonów wapnia, wykazywany przez tetrandrynę i jej pochodną hernandezynę (17). Hamują one napływ do komórki jonów wapnia, wywołany przez wyczerpanie zasobów wapnia wewnątrzkomórkowego śródbłonka naczyń krwionośnych. Jednakże hernandezyna, mająca dodatkową grupę metoksylową, wykazuje słabszą aktywność w porównaniu z tetrandryną.
Inne alkaloidy bis-benzylizochinolinowe. Daurycyna, odkryta w roślinie Menispermum dauricum ( Menispermaceae), odróżnia się od tetrandryny obecnością tylko jednego mostka eterowego między jednostkami benzylizochinolinowymi oraz konformacją β-α. Daurycyna wywiera wpływ blokujący na dokomórkowe prądy sodowe we włóknach Purkynjego w sercu. Tetrandryna jest pozbawiona tego działania. Natomiast antagonizm daurycyny w stosunku do kanałów wapniowych jest słabszy w porównaniu z tetrandryną. Właściwości kliniczne daurycyny są podobne do przeciwarytmicznej chinidyny, należącej do klasy 1a leków antyarytmicznych (5). Daurycyna blokuje także szybkie i wolne prądy potasowe, co w połączeniu z hamowaniem prądu wapniowego czyni z niej prawdopodobny lek antyarytmiczny z minimalnym ryzykiem działań ubocznych w postaci ´Torsades de pointes´(12).
Z tej samej rośliny została wyizolowana dauryzolina, która również wykazuje działanie antyarytmiczne, jeszcze silniejsze w porównaniu z daurycyną. Jej mechanizm działania jest analogiczny do tetrandryny i opiera się na blokowaniu napływu jonów wapnia do komórki przez kanały wapniowe oraz na hamowaniu uwalniania tego jonu z magazynów siateczki endoplazmatycznej (12).
Do alkaloidów bis-benzylizochinolinowych należy także neferyna, otrzymana z rośliny Nelumbo nucifera, ( Nelumbonaceae), mająca jeden mostek eterowy i charakteryzująca się podobnym do daurycyny profilem farmakologicznym (5). Neferyna również zmniejsza ilość wewnątrzkomórkowego wapnia dwutorowo. Została udowodniona jej aktywność jako nieswoistego inhibitora prądu sodowego, wapniowego i potasowego. Suma tych mechanizmów składa się na antyarytmiczne działanie neferyny i jej pozytywny wpływ na układ sercowo-naczyniowy (12).
Biorąc pod uwagę dotychczas publikowane dane, można pokusić się o sformułowanie zależności działania od budowy chemicznej alkaloidów bis-benzylizochinolinowych. Pochodne tetrandryny podstawione w pozycji 7-O różnymi grupami alkilowymi wykazują różnice w działaniu hipotensyjnym. Obecność grupy acetylowej lub demetylacja w tej pozycji powoduje brak działania hipotensyjnego. Pochodne 7-O-etylowe i 7-O-izopropylowe, podobnie jak tetrandryna, wykazują powolny i długotrwały efekt hipotensyjny, bez znacznego wpływu na rytm serca i stężenie reniny w osoczu. Natomiast wstawienie w pozycji 7-O dłuższych łańcuchów, jak n-propylowy lub n-butylowy, redukuje zarówno stopień, jak i długość trwania aktywności hipotensyjnej.
Na aktywność farmakologiczną wywiera również wpływ rodzaj stereoizomerii alkaloidów oraz ilość mostków eterowych obecnych w cząsteczce. Decydują one bowiem o powinowactwie związku do kanału wapniowego. Rezultaty badań wykazały, że wszystkie pochodne tetrandryny całkowicie blokują wiązanie się diltiazemu do receptora w kanale wapniowym, jednakże tetrandryna wykazuje wśród nich największą aktywność. Ponadto niektóre konformery β-α, jak np. daurycyna lub β-β, zawodzą jako stabilizatory wiązania dihydropirydyny, mimo że mają zdolność przyłączania się do receptora benzodiazepinowego w kanale wapniowym.
Powyższe wnioski sugerują, że synteza pochodnych tetrandryny może doprowadzić do otrzymania w przyszłości nowych leków przeciwnadciśnieniowych o znacznej wartości terapeutycznej (5). Sama tetrandryna jest stosowana w Chinach już od ponad pół wieku jako lek hipotensyjny (16).
Alkaloidy indolowe
Alkaloidy indolowe stanowią dużą grupę związków, których wspólną cechą jest występowanie rdzenia indolowego. W zależności od budowy, można podzielić je na związki o stosunkowo prostej budowie, zaliczane do amin lub protoalkaloidów, bardziej złożone, zawierające boczny fragment węglowy oraz alkaloidy dimeryczne.
Alkaloidy indolowe są szeroko rozpowszechnione w świecie roślinnym. Prostsze związki występują m.in. w rodzinach: Passifloraceae, Eleagnaceae i Zygophyllaceae, natomiast alkaloidy o budowie złożonej spotyka się głównie w rodzinach: Apocynaceae, Loganiaceae, Rubiaceae i Calycanthaceae (7).
Hirsutyna. Jednym z alkaloidów indolowych jest hirsutyna, wyodrębniona z rośliny Uncaria rhynchophylla z rodziny Rubiaceae. Jest związkiem o budowie tetracyklicznej (ryc. 3), stosowanym w chińskiej medycynie naturalnej do leczenia bólów głowy i nudności, będących skutkiem nadciśnienia (18).
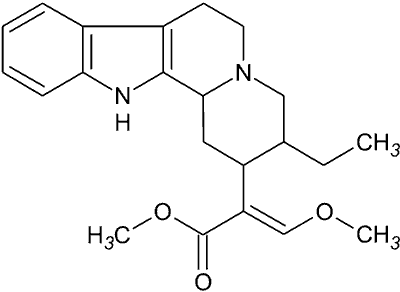
Ryc. 3. Hirsutyna.
Hirsutyna powoduje obniżenie ciśnienia krwi u szczurów i rozluźnia mięśnie gładkie wyizolowanej szczurzej aorty, których skurcz został spowodowany norepinefryną lub roztworem soli potasu o wysokim stężeniu. Sugeruje to, że u podstawy tego efektu leży inhibicja potencjałozależnych kanałów wapniowych (18, 19).
Oprócz hamowania napływu jonów wapnia do komórki przez kanały wapniowe, wywiera także efekt na wapń wewnątrzkomórkowy. Blokuje jego uwalnianie z magazynów wewnątrzkomórkowych oraz zwiększa wychwyt i magazynowanie go w siateczce śródplazmatycznej (20).
Hirsutyna jest nieselektywnym blokerem kanałów jonowych. Stwierdzono także jej zdolność do hamowania prądu sodowego (18). Dzięki zmniejszaniu wewnątrzkomórkowego stężenia jonów wapnia, hirsutyna rozluźnia mięśnie gładkie naczyń krwionośnych, co skutkuje efektem hipotensyjnym. Obniżenie ciśnienia krwi nie powoduje jednakże wtórnej tachykardii, jak dzieje się w przypadku innych, syntetycznych antagonistów wapnia, np. pochodnych dihydropirydyny. Brak efektu ubocznego związany jest z bezpośrednim działaniem hirsutyny na serce. Hirsutyna charakteryzuje się także działaniem antyarytmicznym. Wykazano, że wygasza arytmie spowodowane przez akonitynę u myszy i przez ouabainę u świnek morskich, dzięki blokowaniu prądu sodowego, ale i innych prądów jonowych (18).
Podobne działanie w stosunku do kanałów jonowych wywiera epimer hirsutyny, dihydrokorynanteina, która w porównaniu z hirsutyną ma bardziej płaską strukturę. Różnice w budowie wpływają na działanie na α-adrenoreceptory. Dihydrokorynanteina blokuje ich aktywność, podczas gdy hirsutyna jest tej zdolności pozbawiona. Przeprowadzone badania udowodniły, że planarność cząsteczki jest niezbędna do wystąpienia powinowactwa wobec α-adrenoreceptorów. Jednakże odmienność budowy obu cząsteczek nie ma wpływu na ich zdolność blokowania kanałów jonowych (18).
Dzięki aktywności nieselektywnego blokera kanałów jonowych, hirsutyna wydaję się być obiecującym lekiem hipotensyjnym, nie powodującym niebezpiecznego wtórnego przyspieszenia akcji serca, dzięki aktywności chronotropowo ujemnej i antyarytmicznej. Wymagane są oczywiście dalsze badania nad jej aktywnością farmakologiczną, łącznie z badaniami klinicznymi.
Inne alkaloidy indolowe. Alkaloidy indolowe występują także w korze brazylijskiej rośliny Himalanthus lancifolius, niegdyś znanej jako Plumeria lancifolia, z rodziny Apocynaceae. Sucha kora tej rośliny jest surowcem zielarskim powszechnie stosowanym jako środek przeciwgorączkowy, przeciwzapalny i przeciwbólowy oraz poronny. Wchodzi także w skład leków używanych przy bolesnym miesiączkowaniu oraz osłabiających objawy menopauzy (21).
W korze Himalanthus lancifolius odkryto uleinę i demetoksyaspidosperminę, które należą do alkaloidów indolowych, jednakże brak badań potwierdzających ich aktywność biologiczną. Badaniu została natomiast poddana frakcja alkaloidowa z kory, składająca się głównie z uleiny (62,61%), ale zawierająca również inne, nieznane alkaloidy. Uniemożliwia to stwierdzenie, które ze związków są odpowiedzialne za uzyskane wyniki. Frakcja alkaloidowa kory Himalanthus lancifolius zmienia odpowiedź skurczową mięśni gładkich, co może być skutkiem blokowania napływu jonów wapnia do komórki przez potencjałozależne kanały wapniowe, modyfikacji w mobilizacji wewnątrzkomórkowego wapnia, a nawet zaburzeń w zdolności komórek do wykorzystania jonów wapnia do wywołania skurczu.
In vitro, frakcja alkaloidowa wykazała redukcję maksymalnej odpowiedzi skurczowej w szczurzych aortach, wywołanej przez acetylocholinę i fenylefrynę. Stwierdzono również całkowite rozluźnienie skurczonych krążków aorty, pozbawionych śródbłonka, co wyklucza zaangażowanie eNOS w mechanizm działania.
Dokładne przebadanie frakcji alkaloidowej kory Himalanthus lancifolius oraz zrozumienie mechanizmu działania jej składników może prowadzić do rozwoju nowej grupy środków terapeutycznych, stosowanych w leczeniu schorzeń powiązanych ze wzrostem wewnątrzkomórkowego stężenia jonów wapnia lub z niepożądaną czynnością skurczową mięśni gładkich, takich jak nadciśnienie, bolesne miesiączkowanie i pomenopauzalne zaburzenia przepływu krwi, spowodowane niedoborem estrogenu.
Alkaloidy pochodne pirazyny
Tetrametylpirazyna została wyizolowana w 1957 r. z rośliny Ligusticum wallichii (rodzina Apiaceae). Podziemne części rośliny są tradycyjnym lekiem ziołolecznictwa chińskiego, stosowanym między innymi w celu poprawy krążenia w terapii choroby niedokrwiennej serca i objawów niedokrwienia mózgu, takich jak zawroty głowy, a także w schorzeniach układu sercowo-naczyniowego. Tetrametylpirazyna, inaczej nazywana ligustrazyną, zbudowana jest z pierścienia pirazynowego z czterema symetrycznie usytuowanymi grupami metylowymi (ryc. 4B) (22).
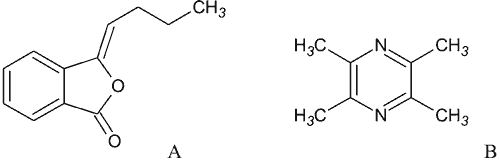
Ryc. 4. Związki czynne Ligusticum wallichi: A – butylidenoftalid, B – tetrametylopirazyna.
Ligustrazyna jest związkiem powodującym silny rozkurcz naczyń krwionośnych płuc, zwłaszcza w warunkach niedotlenienia. Ponadto rozkurcza tętnicę podstawną i krezkową, których skurcz był spowodowany przez podanie KCl, CaCl2 i norepinefryny; zwiększa przepływ nerkowy, przepływ w naczyniówce oka, ogólnoustrojowy i wieńcowy. Wpływ na przepływ wieńcowy został opisany jako krótkotrwały i o niskiej toksyczności. Jednocześnie stwierdzono przyspieszenie rytmu serca. Efekt rozkurczający został także wykazany w stosunku do mięśni gładkich macicy, gdzie omawiana substancja zmniejszała napięcie mięśniowe i hamowała odpowiedź na oksytocynę i prostaglandyny u kobiet ciężarnych (22).
Zarówno w badaniach in vitro, jak i in vivo stwierdzono, że ligustrazyna wykazuje działanie hipotensyjne i bezpośrednio rozkurcza mięśniówkę naczyń krwionośnych na skutek blokowania kanałów wapniowych. Oprócz hamowania napływu jonów wapnia do wnętrza komórki przez kanały wapniowe, hamuje także uwalnianie tych jonów z wewnątrzkomórkowych magazynów mięśni gładkich naczyń krwionośnych. Jest więc prawdziwym i całkowitym antagonistą wapnia (22). Badania nad powinowactwem ligustrazyny do kanałów wapniowych typu L wykazały, że blokuje ona głównie nieaktywne kanały wapniowe. Nie wymaga więc wniknięcia do wnętrza kanału, aby wywierać efekt farmakologiczny.
Ligustrazyna jest szybko wchłaniana po przyjęciu doustnym, ale także szybko wydalana z moczem, więc aby utrzymać jej efekty działania musi być stosowana co kilka godzin. Alternatywnie może być podawana we wlewie kroplowym. W ten sposób stosowana jest w Chinach u chorych hospitalizowanych po ataku serca i zawale (23).
Kolejnym związkiem wyizolowanym z Ligusticum wallichii jest butylidenoftalid (ryc. 4A). Obok innych ftalidów, jest składnikiem olejku eterycznego, otrzymywanego z tej rośliny. Podobnie jak tetrametylpirazyna wykazuje zdolność blokowania potencjałozależnych kanałów wapniowych, jednakże w większym stopniu hamuje uwalnianie jonów wapnia z magazynów wewnątrzkomórkowych. W rezultacie rozluźnia mięśnie gładkie naczyń krwionośnych i ma aktywność przeciwdusznicową, ale nie wywołuje istotnych zmian ciśnienia krwi (24).
Odkrycie mechanizmu działania tetrametylopirazyny i butylidenoftalidu, głównych składników Ligusticum wallichii, potwierdza celowość ich stosowania w schorzeniach układu sercowo-naczyniowego, takich jak choroba niedokrwienna serca, zawał serca, zakrzepica naczyń oraz nadciśnienie tętnicze.
Ginsenozydy
Korzeń żeń-szenia ( Panax ginseng, rodzina Araliaceae, oraz kilka innych gatunków z rodzaju Panax) jest stosowany od tysięcy lat, a jego złożone działanie farmakologiczne było wielokrotnie opisywane w literaturze. Oprócz wielu innych aktywności żeń-szeń wykazuje również ochraniające działanie na układ sercowo-naczyniowy (25).
Za większość efektów farmakologicznych żeń-szenia odpowiedzialne są ginsenozydy, które występują w tej roślinie w postaci ponad dwudziestu różnych związków. Wyjąwszy ginsenozyd Ro, należą one do saponin triterpenowych. Na podstawie budowy aglikonu można podzielić ginsenozydy na dwie grupy: grupę panaksadiolu (np. ginsenozydy Rb1, Rc i Rg3) (ryc. 5) oraz grupę panaksatriolu (np. ginsenozydy Rg1 i Re) (26).
Ryc. 5. Ginsenozyd Rc.
Zdolność do blokowania kanałów wapniowych przez ginsenozydy została wykazana w kilku badaniach (26, 27, 28, 29), a mechanizmy działania zaangażowane w ten efekt są różnorakie.
Po pierwsze, ginsenozydy, wbudowując się w dwuwarstwową błonę komórkową, powodują zmiany aktywności białek błonowych, które budują kanał wapniowy. Spośród pięciu przebadanych ginsenozydów (Rb1, Re, Rf, Rg1 i Rc) największą aktywność hamującą prąd wapniowy wykazał ginsenozyd Rc (29), słabszą zaś ginsenozyd Re (27). Po drugie, zmieniając właściwości dwuwarstwy fosfolipidowej, mogą modulować aktywność białek G, a niektóre kanały wapniowe w neuronach czuciowych są z nimi funkcjonalnie powiązane. Stąd też wynika antynocyceptywna aktywność ginsenozydu Rf (25). Po trzecie, stwierdzono, że ginsenozyd Re hamuje prąd wapniowy typu L przez stymulację wytwarzania NO, który uaktywnia transdukcję sygnału zależną od cGMP (26, 28).
Ginsenozydy wykazują znaczne zróżnicowanie strukturalne, co determinuje odmienność w ich działaniu. Mogą różnić się rodzajem, liczbą i miejscem przyłączenia reszt cukrowych oraz liczbą i miejscem usytuowania podstawnika hydroksylowego. Jest to ważne, gdyż te polarne grupy oddziałują z hydrofilowymi grupami fosfolipidów błony komórkowej, co wpływa na zdolność wstawiania się związku w błonę i wywarcie efektu farmakologicznego (25).
Kolejnym czynnikiem powodującym różnice strukturalne pomiędzy ginsenozydami jest występowanie stereoizomerii przy C-20. Potwierdzeniem są badania nad stereoswoistością epimerów ginsenozydu Rg3, które wykazały, że 20(S)-ginsenozyd Rg3 hamuje prąd wapniowy typu L, podczas gdy drugi epimer, 20(R), jest pozbawiony tej aktywności (29).
Różnorodność ginsenozydów oraz zdolność do wywierania rozmaitych efektów na drodze odmiennych mechanizmów przedstawia te związki jako obiecujące środki lecznicze. Oprócz wielu innych aktywności, mają one zdolność blokowania kanałów wapniowych typu L, dzięki czemu wykazują działanie hipotensyjne (29), antyarytmiczne oraz skracają czas trwania potencjału czynnościowego serca. Chronią także organizm przed skutkami zaburzeń przepływu krwi i niedokrwieniem. Czyni to z nich wartościowe leki, mogące znaleźć zastosowanie w terapii schorzeń układu sercowo-naczyniowego (28).
Izoflawony
Izoflawony, obok lignanów, kumaryn i laktonów kwasu rezorcynowego, należą do fitoestrogenów, czyli substancji pochodzenia roślinnego, strukturalnie i funkcjonalnie podobnych do estradiolu (30). Występują głównie w rodzinie Fabaceae, rzadziej w rodzinach Iridaceae, Rosaceae i in. (7). Powstają jako pochodne flawonów wskutek działania syntaz izoflawonowych (IFS), które przenoszą resztę arylową (pierścień B) z pozycji 2 do pozycji 3 (ryc. 6).
Ryc. 6. Izoflawony: A – genisteina, B – daidzeina, C – ekwol, D – pueraryna.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Olszanecki R., Kocemba J.: Biologiczna rola wapnia-mechanizmy działania i geriatryczne walory leków blokujących kanały wapniowe. Gerontol. Pol. 2004, 12, 133. 2. Rola R.: Potencjałozależne kanały jonowe wapniowe. Med. Dydak. Wychow. 2004, 36, 29. 3. Kwan C.Y., Fi A.: Tetrandrine and related bis-benzylisoquinoline alkaloids from medicinal herbs: cardiovascular effects and mechanisms of action. Acta Pharmacol. Sin. 2002, 23, 1057. 4. Kocurek A., Piwowarska W.: Antagoniści jonów wapnia a skurcz tętnic wieńcowych i zapobieganie restenozie po angioplastyce. Przegl. Lek. 1997, 54, 37. 5. Wang G., Lemos J.R.: Tetrandrine: a new ligand to block voltage-dependent Ca2+ and Ca2+-activated K+ channels. Life Sci. 1995, 56, 295. 6. Sun J., Tan B.K.H., Huang S.H., i wsp.: Effects of natural products on ischemic heart diseases and cardiovascular system´. Acta. Pharmacol. Sin. 2002, 23,1142. 7. Kohlműnzer S.: Farmakognozja. PZWL Warszawa, 2000, 670. 8. Orallo F.: Pharmacological effects of (+)-nantenine, an alkaloid isolated from Platycapnos spicata, in several rat isolated tissues. Planta Med. 2003, 69, 135. 9. Chang G.J., Wu M.H., Wu Y.C. i wsp.: Electrophysiological mechanisms for antiarrhythmic efficacy and positive inotropy of liriodenine, a natural aporphine alkaloid from Fissistigma glaucescens. Br. J. Pharmacol. 1996, 118, 1571. 10. Yao W.X., Jiang M.X.: Effects of tetrandrine on cardiovascular electrophysiologic properties. Acta Pharmacol. Sin. 2002, 23, 1069. 11. Guan S., Lynch C.: Effect of tetrandrine on cellular electrophysiology and calcium uptake of myocardium in guinea pigs and dogs. Chin. Med. J. 2001, 114, 1046. 12. Qian J.Q.: Cardiovascular pharmacological effects of bisbenzylisoquinoline alkaloid derivates. Acta Pharmacol. Sin. 2002, 23, 1086. 13. Wong K.K.: Differential effect of tetrandrine on aortic relaxation and chronotropic activity in rat isolated aorta and atria. Planta Med. 1998, 64, 663. 14. King V.F., Garcia M.L., Himmel D. i wsp.: Interaction of tetrandrine with slowly inactivating calcium channel. J Biol. Chem. 1988, 263, 2238. 15. Wu S., Yu X.C., Shan J. i wsp.: Cardiac effects of the extract and active components of Radix Stephaniae tetrandrae. Electrically-induced intracellular calcium transient and protein release during the calcium paradox. Life Sci. 2001, 68, 2853. 16. Yu X.C., Wu S., Chen C.F. i wsp.: Antihypertensive and antiarrhythmic effects of an extract of Radix Stephaniae tetrandrae in the rat. J. Pharm. Pharmacol. 2004, 56, 115. 17. Low A.M., Berdik M., Sormaz L. i wsp.: Plant alkaloids, tetrandrine and hernandezine, inhibit calcium-depletion stimulated calcium entry in human and bovine endothelial cells. Life Sci. 1996, 58, 2327. 18. Masumiya H., Saitoh T., Tanaka Y. i wsp.: Effects of hirsutine and dihydrocorynantheine on the action potentials of sino-atrial node, atrium and ventricle. Life Sci. 1999, 65, 2333. 19. Yano S., Horiuchi H., Horie S. i wsp.: Ca2+ channel blocking effects of hirsutine, an indole alkaloid from Uncaria genus, in the isolated rat aorta. Planta Med. 1991, 57, 403. 20. Horie S., Yano S., Aimi N. i wsp.: Effects of hirsutine, an antihypertensive indole alkaloid from Uncaria rhynchophylla, on intracellular calcium in rat thoracic aorta. Life Sci. 1992, 50, 491. 21. Rattmann Y.D., Terluk M.R., Souza W.M. i wsp.: Effects of alkaloids of Himatanthus lancifolius (Muell. Arg.) Woodson, Apocynaceae, on smooth muscle responsiveness. J. Ethnopharmacol. 2005, 100, 268. 22. Pang PKT, Shan J.J., Chiu K.W.: Tetramethylpyrazine, a calcium antagonist, Planta Med. 1996, 62, 431. 23. Zou L.Y., Hao X.M., Zhang G.Q. i wsp.: Effect of tetramethyl pyrazine on L-type calcium channel in rat ventricular myocytes. Can. J. Physiol. Pharmacol. 2001, 79, 621. 24. Ko W.C., Chang C.Y., Sheu J.R. i wsp.: Effect of butylidenephthalide on calcium mobilization in isolated rat aorta. J. Pharm. Pharmacol. 1998, 50, 1365. 25. Attele A.S., Wu J.A., Yuan C.S.: Ginseng pharmacology, multiple constituents and multiple actions. Biochem. Pharmacol. 1999, 58, 1685. 26. Scott G.I., Colligan P.B., Ren B.H. i wsp.: Ginsenosides Rb1 and Re decrease cardiac contraction in adult rat ventricular myocytes: role of nitric oxide. Br. J. Pharmacol. 2001, 134, 1159. 27. Jin Z.Q.: The action of ginsenoside Re on inotropy and chronotropy of isolated atria prepared from guinea pigs. Planta Med. 1996, 62, 314. 28. Bai C.X., Takahashi K., Masumiya H. i wsp.: Nitric oxide-dependent modulation of the delayed rectifier K+ current and the L-type Ca2+ current by ginsenoside Re, an ingredient of Panax ginseng, in guinea-pig cardiomyocytes. Br. J. Pharmacol. 2004, 142, 567. 29. Kim J.H., Lee J.H., Jeong S.M. i wsp.: Stereospecific effects of ginsenoside Rg3 epimers on swine coronary artery contractions. Biol. Pharm. Bull 2006, 29, 365. 30. Figtree G.A., Griffiths H., Lu Y.Q. i wsp.: Plant-derived estrogens relax coronary arteries in vitro by a calcium antagonistic mechanism. J. Am. Coll. Cardiol. 2000, 35,1977. 31. Murkies A.L., Wilcox G., Davis S.R.: Clinical review 92, phytoestrogens. J. Clin. Endocrin. Metabol. 1998, 83, 297. 32. Liew R., Williams J.K., Collins P. i wsp.: Soy-derived isoflavones exert opposing actions on guinea pig ventricular myocytes. J. Pharmacol. Experiment Therap. 2003, 304, 985. 33. Ji E., Yue H., Wu Y. i wsp.: Effects of phytoestrogen genistein on myocardial ischemia/reperfusion injury and apoptosis in rabbits. Acta Pharmacol. Sin. 2004, 56, 306. 34. Ji E.S., Yin J.X., Ma H.J. i wsp.: Effect of genistein on L-type calcium current in guinea pig ventricular myocytes. Acta Physiol. Sin. 2004, 56, 466. 35. Chiang C.E., Chen S.A., Chang M.S. i wsp.: Genistein directly inhibits L-type calcium currents but potentiates cAMP-dependent chloride currents in cardiomyocytes. Biochem. Biophys. Res. Comm. 1996, 223, 598. 36. Belevych A.E., Warrier S., Harvey R.D.: Genistein inhibits cardiac L-type Ca2+ channel activity by a tyrosine kinase-independent mechanism. Mol. Pharmacol. 2002, 62, 554. 37. Wang Y.G., Lipsius S.L.: Genistein elicits biphasic effects on L-type Ca2+ current in feline atrial myocytes. Am. J. Physiol. 1998, 275, H204. 38. Ogura T., Shuba L.M., McDonald T.F.: L-type Ca2+ current in guinea pig ventricular myocytes treated with modulators of tyrosine phosphorylation. Am. J. Physiol. 1999, 276, H1724. 39. Ma T., Fan Z.Z., He R.R.: Electrophysiological effects of phytoestrogen genistein on pacemaker cells in sinoatrial nodes of rabbits. Acta Pharmacol. Sin. 2002, 23, 367. 40. Ji E., Wang C., He R.R.: Effects of genistein on intracellular free-calcium concentration in guinea pig ventricular myocytes. Acta Pharmacol. Sin. 2004, 56, 204. 41. Yao R., An J.: Advances in myocardial protection. Internet J. Anesthesiol. 1999, 3, 2. 42. Qian Y., Li Z., Huang L. i wsp.: Blocking effect of puerarin on calcium channel in isolated guinea pig ventricular myocytes. Chin. Med. J. 1999, 112, 787. 43. Rauwald H.W., Brehm O., Odenthal K.P.: The involvement of a Ca2+ channel blocking mode of action in the pharmacology of Ammi visnaga fruits. Planta Med. 1994, 60, 101. 44. Ubeda A., Tejerina T., Tamargo J. i wsp.: Effects of khellin on contractile responses and 45Ca2+ movements in rat isolted aorta. J. Pharm. Pharmacol. 1991, 43, 46. 45. Ubeda A., Villar A.: Relaxant actions of khellin on vascular smooth muscle. J. Pharm. Pharmacol. 1989, 41, 236. 46. Duarte J., Torres A.I., Zarzuelo A.: Cardiovascular effects of visnagin on rats. Planta Med. 2000, 66, 35. 47. Duarte J., Perez-Vizcaino F., Torres A.I. i wsp.: Vasodilator effects of visnagin in isolated rat vascular smooth muscle. Eur. J. Pharmacol. 1995, 286, 115. 48. Duarte J., Vallejo I., Perez-Vizcaino F. i wsp.: Effects of visnadine on rat isolated vascular smooth muscle. Planta Med. 1997, 63, 233. 49. Bodalski T., Karłowicz-Bodalska K.: Ginkgo biloba L. – miłorząb dwuklapowy (chemizm i działanie biologiczne). Post. Fitoter. 2006, 7, 195. 50. Chen B., Cai J., Song L.S. i wsp.: Effects of Ginkgo biloba extract on cation currents in rat ventricular myocytes. Life Sci. 2005, 76, 1111. 51. Qi X.Y., Zhang Z., Xu Y.: Effects of ginkgolide B on action potential and calcium, potassium current in guinea pig ventricular myocytes. Acta Pharmacol. Sin. 2004, 25, 203. 52. Satoh H.: Suppresion of pacemaker activity by Ginkgo biloba extract and its main constituent, bilobalide in rat sino-atrial nodal cells. Life Sci. 2005,78, 67. 53. Sierpina V.S., Wollschlaeger B., Blumenthal M.: Ginkgo biloba. Am. Fam. Physician. 2003, 68, 923. 54. El-Bardai S., Wibo M., Hamaide M.C. i wsp.: Characterisation of marrubenol, a diterpene extracted from Marrubium vulgare, as an L-type calcium channel blocker. Brit. J. Pharmacol. 2003, 140, 1211. 55. Wang G.J., Liao J.F., Hintz K.K. i wsp.: Calcium antagonizing activity of S-petasin, a hypotensive sesquiterpene from Petasites formosanus, on inotropic and chronotropic responses on isolated rat atria and cardiac myocytes. Naunyn-Schmiedeberg´s Arch. Pharmacol. 2004, 369, 322. 56. Zhang Y., Liu Y., Wang T. i wsp.: Resveratrol, a natural ingredient of grape skin: Antiarrhythmic efficacy and ionic mechanisms. Biochem. Biophys. Res. Comm. 2006, 340, 1192. 57. Tsai S.K., Huang S.S., Hong C.Y.: Myocardial protective effect of hinokiol: an active component in Magnolia officinalis, Planta Med. 1996, 62, 503. 58. Lu Y.C., Chen H.H., Ko C.H. i wsp.: The mechanism of hinokiol-induced and magnolol-induced inhibition on muscle contraction and Ca2+ mobilization in rat uterus. Naunyn Schmiedebergs Arch. Pharmacol. 2003, 368, 262. 59. Sugiyama A., Zhu B.M., Takahara A. i wsp.: Cardiac effects of Salvia miltiorrhiza / Dalbergia odorifera mixture, an intravenously applicable Chinese medicine widely used for patients with ischemic heart disease in China. Circ. J. 2002, 66, 182. 60. Fu Y.F., Hok K.J., Ho Y.J. i wsp.: Pharmacological evidence for calcium channel inhibition by Danshen ( Salvia miltiorrhiza) on rat isolated femoral artery. J. Cardiovasc. Pharmacol. 2006, 47, 139.