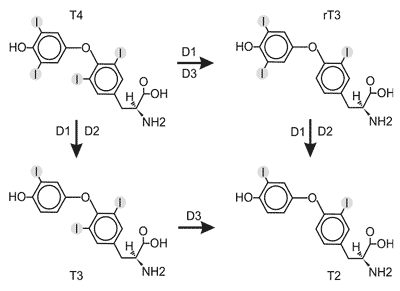
Dejodynaza typu I była pierwszym odkrytym enzymem katalizującym konwersję T4 do T3 (10,11), pierwsza też została sklonowana (6, 12). D1 jako jedyna spośród dejodynaz, katalizuje reakcję odjodowania w pozycji 5´ (z wytworzeniem T3 i T2), jak i w pozycji 5 (z wytworzeniem rT3). D1 wykazuje najwyższe powino-wactwo do rT3, mniejsze do T4 i T3 (rT3<
W stanie eutyreozy D1 odpowiada za syntezę około 34% obwodowej T3, natomiast w stanie tyreotoksykozy jej udział wzrasta do 67% (i więcej) w zależności od stopnia zaawansowania choroby (16). Aktywność D1 specyficznie hamuje 6-n-propylotiouracyl (PTU); właściwość ta jest wykorzystywana do różnicowania aktywności D1 od pozostałych dejodynaz.
Gen kodujący D1 ( Dio1) w genomie ludzkim zlokalizowany jest w pozycji 1p32-33, składa się z 4 eksonów (17). Transkrypcja Dio1jest regulowana między innymi przez hormony tarczycy. W promotorze Dio1znajdują się dwa pozytywne TRE: klasycznyTRE-DR4 (18,19) oraz nietypowy TRE-DR10 (18), umożliwiające aktywację transkrypcji genu przez receptory TR, a także miejsca wiązania czynnika transkrypcyjnego Sp1 (19, 20).
Pierwotny transkrypt Dio1podlega procesowi różnicowego składania eksonów, w wyniku którego powstają warianty transkrypcyjne o różnej sekwencji, w tym pozbawione elementów kodujących rejon białka zawierający centrum aktywne (ryc. 4).
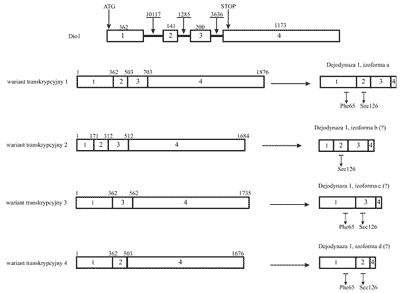
Ryc. 4. Gen, transkrypt i produkt białkowy ludzkiej dejodynazy 1. Rycina przedstawia cztery istniejące w bazie danych Entrez-Gene warianty splicingowe i powstające na ich matrycy produkty białkowe. W literaturze opisano jedynie wariant 1 i odpowiadającą mu izoformę a. Znakiem "?” oznaczono izoformy dejodynazy, których istnienie in vivo nie zostało jeszcze potwierdzone. Eksony zaznaczono prostokątami i ponumerowano od 1 do 4. Liczby ponad eksonami i intronami wskazują na ich wielkość w parach zasad. Na schemacie genu zaznaczono pozycje kodonów ATG i STOP. Na schemacie białka zaznaczono aminokwasy istotne z punktu widzenia aktywności enzymatycznej dejodynazy 1.
Ekspresja dejodynazy 1 w tkankach nowotworowych
Ekspresja genu Dio1jest zaburzona w nowotworach. Poziom mRNA i aktywności D1 są obniżone w raku brodawkowatym tarczycy (21-24), w raku jasnokomórkowym nerki (25, 26), w guzach glejowych mózgu (27), podwyższony w gruczolaku pęcherzykowym i raku pęcherzykowym tarczycy (21), raku piersi (28). Badania na liniach komórkowych raka piersi MCF-7 i MDA-MB-231 sugerują, że podwyższona ekspresja D1 ( Dio1) może stanowić marker różnicowania w komórkach nowotworowych (29).
Pierwsze doniesienia dotyczące zaburzonej ekspresji D1 w ccRCC zostały opublikowane przez nasz zespół w 1991 r. (25). Stwierdzono wówczas, że aktywność D1 była znacząco obniżona w tkance nowotworowej w porównaniu ze stanowiącą kontrolę zdrową tkanką otaczającą guz. Obniżona aktywność D1 korelowała z obniżonym poziomem białka. Kolejne badania wykazały znaczne zróżnicowanie aktywności D1 oraz jej ekspresji na poziomie mRNA pomiędzy próbkami kontrolnymi pobranymi od różnych pacjentów. Różnice w poziomie mRNA były nawet 10-krotne. W tkankach nowotworo-wych zarówno aktywność enzymatyczna jak i ilość mRNA D1 były prawie niewykrywalne (30, 26). Jedną z przyczyn obniżonej ekspresji D1 w nowotworach nerki może być zaburzona aktywacja transkrypcji Dio1przez receptory TR. Zmutowane wersje TR sklonowane z tkanek ccRCC aktywowały promotor genu Dio1znacznie słabiej, niż dzikie typy tych (31).
Analiza ekspresji Dio1na poziomie posttranskrypcyjnym
Analiza wariantów transkrypcyjnych klonowanych z tkanek ccRCC oraz z tkanek nienowotworowych z przeciwległego bieguna nerki nowotworowej, jak również z próbek pobranych z nerek nienowotworowych (powypadkowych, kamiczych, ektopowych, marskich) wykazała istnienie wielu nieopisanych do tej pory wariantów transkrypcyjnych Dio1, w tym wariantów pozbawionych eksonu 2, zawierającego rejon kodujący centrum aktywne. Zidentyfikowano m.in. warianty zawierające wszystkie 4 eksony, eksony: 1, 2, 4, eksony: 1 i 4, a nawet wariant składający się tylko z eksonu 1. Dokładna analiza poziomu ekspresji transkryptów genu Dio1,wykonana za pomocą techniki PCR czasu rzeczywistego ( real-time PCR) wykazała dramatycznie obniżony poziom ekspresji wszystkich wariantów transkrypcyjnych (o około 90%) w tkankach nowotworowych w porównaniu z tkankami kontrolnymi (32). Zaburzony był również proces różnicowego składania eksonów. W tkankach nowotworowych stwierdzono istotny statystycznie spadek stosunku wariantów transkrypcyjnych zawierających rejon kodujący centrum aktywne białka i wariantów pozbawionych tego rejonu. Stwierdzono, że zaburzenia różnicowego składania eksonów D1 wynikają prawdopodobnie z nieprawidłowej ekspresji czynników splicingowych SF2/ASF i hnRNPA1 w ccRCC (Piekiełko-Witkowska A., wyniki niepublikowane).
Analiza ekspresji receptorów jądrowych trijodotyroniny
Receptory jądrowe trijodotyroniny, TR, to średniej wielkości białka jądrowe (TRβ1- 461 aminokwasów; 53 kDa), kodowane przez geny zlokalizowane na chromosomach trzecim ( THRB - 3p24.2) i siedemnastym ( THRA - 17q23).
W procesie różnicowego składania pierwotnego transkryptu powstają po dwie główne izoformy każdego receptora – odpowiednio TRα1 i TRα2 oraz TRβ1 i TRβ2. Receptory jądrowe T3 wpływają na ekspresję genów docelowych poprzez selektywne wiązanie się z promotorowymi sekwencjami DNA typu HRE ( Hormone Response Element), wzmacniając tym samym (lub osłabiając) wydajność transkrypcji. TR, podobnie jak inne białka z rodziny receptorów jądrowych, wykazują obecność dobrze scharakteryzowanych domen funkcjonalnych (ryc. 5). Wiązanie hormonu powoduje zmiany konformacyjne białka powodując zmiany w oddziaływaniu TR z koaktywatorami i korepresorami.
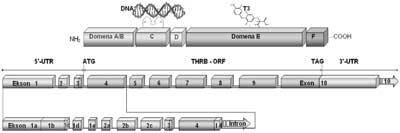
Ryc. 5. Schemat budowy domenowej białka receptorowego dla trijodotyroniny TRβ1 (A.) oraz układu eksonów transkryptu THRB (B.).
W budowie receptora TRβ1 wyróżniono: domenę A/B, odpowiedzialną za wiązanie białek regulatorowych oraz niezależną od T3 aktywację transkrypcji, domenę C wiążącą DNA (8Cys=2 palce cynkowe), domenę łącznikową D zawierającą sygnał lokalizacji jądrowej, domenę E odpowiedzialną za wiązanie hormonu (T3) i dimeryzację receptorów oraz domenę F odpowiedzialną za zależną od T3 aktywację transkrypcyjną receptora.
Na schemacie przedstawiono ponumerowane kolejno eksony transkryptu THRB (B.) z wyszczególnieniem eksonów 5´UTR (1a, 1b, 1c, 1d, 1e, 2a, 2b, 2c) powstających w procesie różnicowego składania pierwotnego transkryptu do alternatywnych form mRNA, różniących się sekwencją obszaru regulatorowego nie ulegającego translacji. Na schemacie zaznaczono również kodon startu translacji, kodon zamknięcia ramki odczytu (TAG) oraz sekwencję kodującą (THRB-ORF).
Regulacja pretranskrypcyjna ekspresji TR w jasnokomórkowym raku nerki: regulacja poprzez wiązanie czynników transkrypcyjnych - receptorów trijodotyroniny (TR) z regulatorAmi sekwencjami promotorowymi (TRE)
W pracy dotyczącej ekspresji TR w raku jasnokomórkowym nerki (33) stwierdzono, że ilość mRNA TRα1 była znamiennie zmniejszona w większości przypadków ccRCC w porównaniu z odpowiadającymi im tkankami kontrolnymi, natomiast ekspresja TRβ1 na poziomie mRNA była bardziej heterogenna: w 70% raków była znacząco zmniejszona, zaś w 30% – zwiększona w guzach w porównaniu z odpowiadającymi im kontrolami. Średnia ilość białka TRα1 była wyższa w ccRCC w porównaniu z tkankami kontrolnymi, podczas, gdy średnia ekspresja TRβ1 na poziomie białka była obniżona w tkankach tych samych nowotworów. Zwiększenie ilości TR niekoniecznie przekładało się na zwiększenie aktywności genów docelowych trijodotyroniny, ponieważ funkcja TR może być, i zwykle jest, zaburzona wskutek różnych mechanizmów, na przykład mutacji w genach kodujących TR, albo, prawdopodobnie, zaburzeń w modyfikacjach potranslacyjnych białka. W ccRCC znaleziono mutacje w receptorze trijodotyroniny (34). Mutacje dotyczyły obu izoform TRα1 i TRβ1; większość z nich znajdowała się w domenie wiążącej ligand, kilka zaś w domenie wiążącej DNA. Ponadto, odsetek tkanek ccRCC, w których stwierdzono mutacje TR wzrastał wraz ze spadkiem zróżnicowania histologicznego nowotworu (34). Badania funkcjonalne in vitro mutantów TR wykazały, że w zależności od miejsca, gdzie doszło do zmiany aminokwasu, mutanty takie nieprawidłowo wiążą T3 lub/i DNA oraz w sposób niedostateczny aktywują geny docelowe (34-38).
Regulacja ekspresji THRB na poziomie potranskrypcyjnym
Składanie pre-mRNA ( splicing) THRB (ryc. 5B) polega na usuwaniu z pierwotnego transkryptu preRNA sekwencji niekodujących (intronów) i łączeniu pozostałych eksonów w gotowy do translacji mRNA. W wyniku alternatywnego składania mogą powstawać kodowane przez ten sam gen białka, różniące się od siebie sekwencją aminokwasów i funkcją, jak również białka identyczne, chociaż kodowane przez mRNA różniące się sekwencją UTR ( UnTranslated Region). UTR to część mRNA nie ulegająca translacji, położona w kierunku 5´ (5´-UTR) lub 3´ (3´-UTR) od sekwencji kodującej. Procesowi składania pre-mRNA podlegają również wspomniane obszary mRNA (5´- oraz 3´-UTR), których funkcja w dojrzałym mRNA polega na regulacji ekspresji genu na poziomie pre-translacyjnym. Najnowsze badania wskazują, że poziom translacji wariantów TR może być regulowany przez takie rejony mRNA (39). Innymi słowy, ilość powstającego białka TR jest zależna od wariantu splicingowego regionu 5´UTR mRNA. Obszary mRNA nie ulegające translacji mogą wpływać na trwałość cząsteczki RNA jak również inicjację oraz efektywność przebiegu samej translacji przez oddziaływanie obszarów 5´UTR na sekwencję kodującą, na tworzenie struktur II-rzędowych utrudniających inicjację translacji (40-41) jak również poprzez wiązanie małych cząsteczek regulatorowych miRNA do sekwencji 3´UTR, obniżających najczęściej efektywność procesu translacji (42-46). W ostatnim czasie wskazuje się na rolę miRNA oraz sekwencji 3´UTR w patogenezie nowotworów (46-48). Sekwencje mRNA nie ulegające translacji stanowią część znaczącego mechanizmu regulacji ekspresji genów na poziomie pre-translacyjnym.
Analiza ekspresji produktów alternatywnego składania THRB
Badania miały na celu określenie struktury i funkcji produktów różnicowego składania niekodujących sekwencji UTR pierwotnego transkryptu receptora TRβ1 w aspekcie jego ekspresji w prawidłowych i nowotworowo zmienionych komórkach. Analizowano ekspresję alternatywnie składanych form transkryptu izoformy receptora jądrowego trijodotyroniny TRβ1 w obrębie sekwencji nie ulegających translacji 5´-UTR. Ustalenie profilu form 5´-UTR wykonano w oparciu o łańcuchową reakcję polimerazy, z wykorzystaniem par starterów flankujących sekwencję niekodującą 5´UTR (w kierunku „forward”): ekson 1a,1b,1c lub 2a i sekwencję kodującą (w kierunku „reverse”): ekson 4 (początek ramki odczytu) lub ekson 10 (koniec ramki odczytu). Analizie jakościowej poddano pary kontrola-ccRCC, ustalone linie komórkowe różnego pochodzenia. Caki-2 (ccRCC), Caki-1 (przerzut skórny ccRCC), HK2 (kanaliki proksymalne nerki ludzkiej), HEK 293 (ludzkie komórki embrionalne nerki), MCF-7 (rak piersi), Jurkat cell line (ludzkie komórki limfoblastyczne T), Raji cell line (chłoniak B-komórkowy) jak również prawidłowe komórki nabłonka jamy ustnej oraz prawidłowe komórki jądrzaste krwi.
Analiza profili form 5´-UTR THRB wykazała, że: ekspresja alternatywnie składanych form splicingowych sekwencji 5´UTR THRB jest zależna od rodzaju tkanki i zaburzona w ccRCC. Prowadzi to do powstania różnych profili 5´UTR w ccRCC i kontrolach. Analiza profili 5´UTR wykazała zaburzenia alternatywnego splicingu nie tylko w ccRCC, ale również w ich kontrolach z przeciwległego bieguna nerki (różne profile eksonów 1a, 1b, 1c 5´UTR między kontrolami), co można próbować tłumaczyć parakrynnym i endokrynnym oddziaływaniem komórek ccRCC na tkankę otaczającą guz.
Analiza porównawcza profili wykazała, że w przeciwieństwie do wszystkich badanych eksonów THRB (1a, 1b, 1c), ekson 2a charakteryzuje się wysoką stabilnością profilu (układu prążków) nie tylko między różnymi tkankami, ale również między ccRCC oraz ich kontrolami, sugerując duże znaczenie tego eksonu dla ekspresji białka, jak również stabilność systemu różnicowego składania pierwotnego transkryptu dla eksonu 2 genu THRB.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Oetting A, Yen PM: New insights into thyroid hormone action. Best Pract Res Clin Endocrinol Metab 2007; 21: 193-208.
2. Baqui M, et al.: Human type 3 iodothyronine selenodeiodinase is located in the plasma membrane and undergoes rapid internalization to endosomes. J Biol Chem 2003; 278: 1206-11.
3. Baqui MM, et al.: Distinct subcellular localization of transiently expressed types 1 and 2 iodothyronine deiodinases as determined by immunofluorescence confocal microscopy. Endocrinology 2000; 141: 4309-12.
4. Curcio-Morelli C, et al.: In vivo dimerization of types 1, 2, and 3 iodothyronine selenodeiodinases. Endocrinology 2003; 144: 937-46.
5. Simpson GI, Leonard DM, Leonard JL: Identification of the key residues responsible for the assembly of selenodeiodinases. J Biol Chem 2006; 281: 14615-21.
6. Berry MJ, Banu L, Larsen PR: Type I iodothyronine deiodinase is a selenocysteine-containing enzyme. Nature 1991; 349: 438-40.
7. Croteau W, et al.: Cloning of the mammalian type II iodothyronine deiodinase. A selenoprotein differentially expressed and regulated in human and rat brain and other tissues. J Clin Invest 1996; 98: 405-17.
8. St Germain DL, et al.: A thyroid hormone-regulated gene in Xenopus laevis encodes a type III iodothyronine 5-deiodinase. Proc Natl Acad Sci U S A 1994; 91: 7767-71.
9. Bianco AC, et al.: Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr Rev 2002; 23: 38-89.
10. Visser TJ, et al.: Subcellular localization of a rat liver enzyme converting thyroxine into tri-iodothyronine and possible involvement of essential thiol groups. Biochem J 1976; 157: 479-82.
11. Hesch RD, Brunner G, Soling HD: Conversion of thyroxine (T4) and triiodothyronine (T3) and the subcellular localisation of the converting enzyme. Clin Chim Acta 1975; 59: 209-213.
12. Mandel SJ, et al.: Cloning and in vitro expression of the human selenoprotein, type I iodothyronine deiodinase. J Clin Endocrinol Metab 1992; 75: 1133-9.
13. Köhrle J: Local activation and inactivation of thyroid hormones: the deiodinase family. Mol Cell Endocrinol 1999; 151: 103-19.
14. Koenig RJ: Regulation of type 1 Iodothyronine deiodinase in health and disease. Thyroid 2005; 15: 835-40.
15. Leonard JL, et al.: Localization of type I iodothyronine 5´-deiodinase to the basolateral plasma membrane in renal cortical epithelial cells. J Biol Chem 1991; 266: 11262-9.
16. Maia AL, et al.: Type 2 iodothyronine deiodinase is the major source of plasma T3 in euthyroid humans. J Clin Invest 2005; 115: 2524-33.
17. Jakobs TC, et al.: Structure of the human type I iodothyronine 5´-deiodinase gene and localization to chromosome 1p32-p33. Genomics 1997; 42: 361-3.
18. Toyoda N, et al.: A novel retinoid X receptor-independent thyroid hormone response element is present in the human type 1 deiodinase gene. Mol Cell Biol 1995; 15: 5100-12.
19. Jakobs TC, et al.: The promoter of the human type I 5´-deiodinase gene-mapping of the transcription start site and identification of a DR+4 thyroid-hormone-responsive element. Eur J Biochem 1997; 247: 288-97.
20. Zhang CY, et al.: Further characterization of thyroid hormone response elements in the human type 1 iodothyronine deiodinase gene. Endocrinology 1998; 139: 1156-63.
21. de Souza Meyer EL, et al.: Decreased type 1 iodothyronine deiodinase expression might be an early and discrete event in thyroid cell dedifferentation towards papillary carcinoma. Clin Endocrinol (Oxf) 2005; 62: 672-8.
22. Ambroziak M, et al.: Disturbed expression of type 1 and type 2 iodothyronine deiodinase as well as titf1/nkx2-1 and pax-8 transcription factor genes in papillary thyroid cancer. Thyroid 2005; 15: 1137-46.
23. Arnaldi LA, et al.: Gene expression profiles reveal that DCN, DIO1, and DIO2 are underexpressed in benign and malignant thyroid tumors. Thyroid 2005; 15: 210-21.
24. Huang Y, et al.: Gene expression in papillary thyroid carcinoma reveals highly consistent profiles. Proc Natl Acad Sci U S A 2001; 98(26): 15044-9.
25. Nauman A, et al.: Aktywność T4-5´-dejodynazy w raku jasnokomórkowym nerki. Urologia Polska Supp. 1991; 339-342.
26. Pachucki J, et al.: Type I 5´-iodothyronine deiodinase activity and mRNA are remarkably reduced in renal clear cell carcinoma. J Endocrinol Invest 2001; 24: 253-61.
27. Nauman P, et al.: The concentration of thyroid hormones and activities of iodothyronine deiodinases are altered in human brain gliomas. Folia Neuropathol 2004; 42: 67-73.
28. Debski MG, et al.: Human breast cancer tissue expresses high level of type 1 5´-deiodinase. Thyroid 2007; 17: 3-10.
29. Garcia-Solis P, Aceves C: 5´Deiodinase in two breast cancer cell lines: effect of triiodothyronine, isoproterenol and retinoids. Mol Cell Endocrinol 2003; 201: 25-31.
30. Nauman A, i wsp.: Aktywność T4-5´-dejodynazy w raku jasnokomórkowym nerki. Urologia Polska Supp. 1991; 339-342
31. Pietrzak M, et al.: 5´-deiodinase type I gene (hdioI) is abnormally activated by thyroid hormone receptor mutants cloned from human kidney cancers. European Journal of Biochemistry 2003; Supplement, abstract number 1.2 P30.
32. Piekielko-Witkowska A, et al.: The expression of alternatively spliced forms of type 1 deiodinase is changed in clear cell renal cell carcinoma. European Congress of Endocrinology 2007; Endocrine Abstracts 2007, 14 P119.
33. Puzianowska-Kuznicka M, et al.: Expression of thyroid hormone receptors is disturbed in human renal clear cell carcinoma. Cancer Lett 2000; 155: 145-52.
34. Kamiya Y, et al.: Expression of mutant thyroid hormone nuclear receptors is associated with human renal clear cell carcinoma. Carcinogenesis 2002; 23: 25-33.
35. Lin KH, et al.: Expression of mutant thyroid hormone nuclear receptors in human hepatocellular carcinoma cells. Mol Carcinog 1999; 26: 53-61.
36. Lin KH, Wu YH, Chen SL: Impaired interaction of mutant thyroid hormone receptors associated with human hepatocellular carcinoma with transcriptional coregulators. Endocrinology 2001; 142: 653-62.
37. Puzianowska-Kuznicka M, et al.: Functionally impaired thyroid hormone receptor mutants are present in thyroid papillary cancer. J Clin Endocrinol Metab 2002; 87: 1120-8.
38. Chan IH, Privalsky ML.: Thyroid hormone receptors mutated in liver cancer function as distorted antimorphs. Oncogene 2006; 25: 3576-88.
39. Frankton S, et al.: Multiple messenger ribonucleic acid variants regulate cell-specific expression of human thyroid hormone receptor beta1. Mol Endocrinol. 2004 18:1631-42.
40. Frankton S, et al.: Multiple messenger ribonucleic acid variants regulate cell-specific expression of human thyroid hormone receptor beta1. Mol Endocrinol. 2004;18(7):1631-42.
41. Hughes TA: Regulation of gene expression by alternative untranslated regions. Trends Genet. 2006;22(3):119-22.
42. Shyu AB, Wilkinson MF, van Hoof A: Messenger RNA regulation: to translate or to degrade. EMBO J. 2008;27(3):471-81.
43. Kozak M: How strong is the case for regulation of the initiation step of translation by elements at the 3´ end of eukaryotic mRNAs? Gene. 2004 Dec 8;343(1):41-54.
44. Filipowicz W, Bhattacharyya SN, Sonenberg N: Mechanisms of post-transcriptional regulation by microRNAs: are the answers insight? Nat Rev Genet. 2008;9(2):102-14.
45. Jaskiewicz L, Filipowicz W: Role of Dicer in posttranscriptional RNA silencing. Curr Top Microbiol Immunol. 2008; 320:77-97.
46. Morris KV. RNA-mediated transcriptional gene silencing in human cells. Curr Top Microbiol Immunol. 2008; 320:211-24.
47. Barbarotto E, Schmittgen TD, Calin GA: MicroRNAs and cancer: profile, profile, profile. Int J Cancer. 2008;122(5):969-77.
48. Cowland JB, Hother C, Grřnbaek K: MicroRNAs and cancer. APMIS. 2007 115(10):1090-106.
49. Skaftnesmo KO, et al.: MicroRNAs in tumorigenesis. Curr Pharm Biotechnol. 2007 8(6):320-5.
50. Master A, et al.: The expression of alternatively spliced 5´-UTR mRNA variants of human thyroid hormone receptor beta 1 (THRB1) is dependent on the tissue type and aberrant in clear cell renal cell carcinoma (ccRCC). Hormone Research, 2007; 48-49.
51. Bedrosian I, et al.: Deregulation alters the biologic properties of ovarian cancer cells. Oncogene 2004; 23: 2648-2657.
52. Nauman A, et al.: Elevated cyclin E level in human clear cell Renal Cell carcinoma: possible causes and consequences. Acta Biochimica Polonica 2007; 54: 595-602.
53. Bieda M, et al.: Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res 2006; 16: 595-605.
54. Pierce AM, et al.: E2F1 has both oncogenic and tumor-suppressive properties in a transgenic model. Mol Cell Biol 1999; 19: 6408-14.
55. DeGregori J, et al.: Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci USA 1997; 94: 7245-7250.
56. Hallstrom TC, Nevins JR: Specificity in the activation and control of transcription factor E2F-dependent apoptosis. Proc Natl Acad Sci USA 2003; 100: 10848-53.
57. Sherr CJ. Cancer cell cycles. Science 1996; 274: 1672-7.
58. Geng Y, et al.: Regulation of cyclin E transcription by E2Fs and retinoblastoma protein. Oncogene 1996; 12: 1173-80.
59. Helin K: Regulation of cell proliferation by the E2F transcription factors. Curr Opin Genet Dev 1998; 8: 28-35.
60. Moller MB, et al.: Frequent disruption of the RB1 pathway in diffuse large B cell lymphoma: prognostic significance of E2F-1 and p16INK4A. Leukemia 2000; 14: 898-904.
61. Turowska O, et al.: Overexpression of E2F1 in Clear Cell Renal Cell Carcinoma: A Potential Impact of Erroneous Regulation by Thyroid Hormone Nuclear Receptors. Thyroid 2007; 17: 1039-48.
62. Poplawski P, Nauman A: triiodothyronine leads to increased proliferation of clear cell renal cell carcinoma cells by changing expression of retinoblastoma family proteins EMBO Workshop 2007 „Molecular Mechanisms of cell cycle control in normal and malignant cells”, abstrakt nr 63.
63. Nicol D, et al.: Vascular endothelial growth factor expression is increased in renal cell carcinoma. J Urol 1997; 157(4): 1482-6.
64. Djordjevic G, et al.: Prognostic significance of vascular endothelial growth factor expression in clear cell renal cell carcinoma. Pathol Res Pract 2007; 203(2): 99-106.
65. Gnarra JR, et al.: Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet 1994; 7: 85-90.
66. van den Berg A, et al.: Analysis of multiple renal cell adenomas and carcinomas suggests allelic loss at 3p21 to be a prerequisite for malignant development. Genes Chromosomes Cancer 1997; 19: 228-32.
67. Pietrzykowski A: Receptory jądrowe dla trijodotyroniny (TRβα1 i TRβ1) i dla kwasu retinowego (RARβ) w raku jasnokomórkowym nerki ludzkiej. Rozprawa doktorska (1997), (Biblioteka CMKP).
68. Nauman A, et al.: The low T3 syndrom in patients with kidney cancer – effect of cancer differentiation. Polish J Endocrinol 1996; 47: 365-74.
69. Tański Z: Zespół niskiej trijodotyroniny u chorych na raka nerki zależność od rozmiaru guza i stopnia jego zróżnicowania. Urologia Polska 2000; 53/3.
70. De Groot L: Dangerous dogmas in medicine: the nonthyroidal illness syndrome. J Clin Endocrinol Metab 1999, 84: 151-164.