© Borgis - Postępy Nauk Medycznych 8/2008, s. 519-525
*Aleksandra Tołoczko-Grabarek, Joanna Matyjasik, Jan Lubiński
Genetyka kliniczna nowotworów nerek
Clinical genetic of kidney cancer
Międzynarodowe Centrum Nowotworów Dziedzicznych Zakład Genetyki i Patomorfologii Pomorska Akademia Medyczna, Szczecin
Kierownik Zakładu: prof. dr hab. med. Jan Lubiński
Streszczenie
Wśród nowotworów nerki można wyodrębnić 2 główne grupy:
1. guz Wilmsa- nephroblastoma
2. raki nerki-adenocarcinoma (carcinoma clarocellulare, carcinoma papillare, colecting duct carcinoma, chromophobic cell carcinoma), carcinoma urotheliale.
Postać rodzinna guza Wilmsa stanowi 1-2% przypadków tych guzów.
Dotychczas opisano 14 genetycznie uwarunkowanych zespołów związanych ze zwiększonym ryzykiem guza Wilmsa. Mutacje konstytucyjne będące przyczyną powstawania dziedzicznego nephroblastoma dotyczą najczęściej jednego z czterech genów: WT1, WT2, FWT1, FWT2. Ryzyko zachorowania na guza Wilmsa u nosicieli tych mutacji wynosi 5% - 90% w zależności od zmutowanego genu.
Opisano 19 zespołów dziedzicznej predyspozycji do nowotworów, w przebiegu których w nerkach mogą powstać rak jasnokomórkowy (CCRC), rak brodawkowaty (PRCC) lub rak z nabłonka przejściowego. Najczęściej rozpoznawanym zespołem jest rodzinny rak jasnokomórkowy nerki (F-CCRC). Wśród wszystkich rodzin CCRC 5% stanowią rodziny spełniające kryteria definitywne F-CCRC, zaś 13% stanowią rodziny CCRC-u kolejnych osób.
W większości przypadków F-CCRC opisanych w literaturze nie znaleziono mutacji będących przyczyną agregacji CCRC w rodzinach. W naszych Ośrodku wykazaliśmy, że istotną przyczyną powstawania CCRC jest konstytucyjna zmiana w genie CHEK2. Nadal jednak kluczową rolę w rozpoznawaniu F-CCRC odgrywa analiza rodowodowo-kliniczna. W naszym Ośrodku w rodzinach z pojedynczym CCRC wysuwamy podejrzenie F-CCRC stosując jako kryteria:
a) zdiagnozowanie CCRC poniżej 55 r.ż. lub
b) wystąpienie raka żołądka lub płuca u krewnych I stopnia pacjenta z CCRC.
U osób z rodzin z rozpoznanym F-CCRC stosujemy schemat badań kontrolnych pozwalający na wczesne wykrycie raka nerki, jednak postepowanie lecznicze nie jest ściśle określone.
Ewentualne schematy leczenia będą mogły być ustalone po analizie przebiegu klinicznego leczonych w różny sposób dużych grup raków z rodzin z F-CCRC.
Summary
Among kidney malignancies two main groups can be distinguished:
1. Wilm´s tumor – nephroblastoma
2. Kidney cancer – adenocarcinoma (carcinoma clarocellulare - CCRC, carcinoma papillare - PRCC, collecting duct carcinoma, chromophobic cell carcinoma), carcinoma urotheliale.
Wilm´s tumor and kidney cancer can develop as result of strong hereditary predisposition.
Familial Wilm´s tumor constitutes 1-2% of all.
Fourteen different syndromes with increased risk of Wilm´s tumor have been reported. Hereditary Wilm´s tumor is caused by constitutional mutation in WT1, WT2, FWT1 or FWT2 genes. Depending on mutated gene, the risk of Wilm´s tumor varies between 5% and 90%.
Kidney cancer can appear in 19 different hereditary cancer syndromes. One of the most often diagnosed is familial clear cell renal cancer syndrome (F-CCRC). Based on our results, in 5% we can diagnose F-CCRC, and 13% are families with only one affected by CCRC but with high probability of CCRC in the relatives.
We found that mutations in CHEK2 are associated with increased risk of CCRC, however in most cases genes causing CCRC and F-CCRC syndrome have been not identified and still the pedigree and clinical data are crucial for appropriate diagnosis.
Results of our study of CCRC patients revealed that F-CCRC syndrome could be recognize if:
a) CCRC was diagnosed before the age 55 y.
b) Io relatives of CCRC patient were affected by stomach or lung cancers.
In all individuals with recognized F-CCRC syndrome, specific surveillance is recommended.
Medical treatment for CCRC patients with F-CCRC is not defined yet, and should be determined.

GUZ WILMSA
Guz Wilmsa ( nephroblastoma) jest najczęstszym złośliwym guzem nerek wieku dziecięcego. Występuje u ok. 1:10 tys. dzieci poniżej 15 roku życia (1). Większość przypadków występuje sporadycznie tj. jako pojedyncze zachorowanie w rodzinie. Postać rodzinna guza Wilmsa stanowi ok. 1-2% przypadków tych guzów.
Genetyczne uwarunkowania guza Wilmsa
Mutacje konstytucyjne będące przyczyną powstawania dziedzicznego nephroblastoma dotyczą najczęściej jednego z czterech genów: WT1, WT2, FWT1, FWT2 (2, 3).
W ok. 10% przypadków u pacjentów z nephroblastoma występują wnętrostwo, spodziectwo, dysmorfie twarzy lub zespoły wad rozwojowych: BWS, WAGR, DDS (tab. 1).
Tabela 1. Genetycznie uwarunkowane zespoły związane ze zwiększonym ryzykiem guza Wilmsa.
| Zespół | Fenotyp | Gen/locus | Dziedziczenie | Ryzyko guza Wilmsa |
| Rodzinna agregacja guzów Wilmsa bez towarzyszących zmian klinicznych | Agregacja rodzinna guzów Wilmsa; guzy częściej jednostronne | WT1(11p13)
WT2(11p15)
FWT1(17q)
FWT2(19q) | AD | Mutacje genów WT1,WT2- ~25%; mutacje genu FWT1-30%; mutacje genu FWT2-70% |
| Denysa-Drasha(DDS) | Guz Wilmsa, glomerulopatia, obojnactwo rzekome | Mutacja punktowa WT1 (11p13) | Mutacje germinalne de novo, rzadko agregacja rodzinna, AD | 90% |
| WAGR | Guz Wilmsa, aniridia, wady układu moczowo-płciowego, upośledzenie umysłowe | Delecja genu WT1(11p13) | Mutacje germinalne de novo, rzadko agregacja rodzinna, AD | 30% |
| Beckwith-Wiedemanna (BWS) | Macrosomia, przerost języka, wady przedniej ściany jamy brzusznej
Zmienne objawy: visceromegalia, hipoglikemia, przerost połowiczy, wady układu moczowo-płciowego, nowotwory embrionalne | Locus WT2 (11p15) | Mutacje germinalne de novo, w 15% rodzinny BWS, AD | 5% |
| Simpsona-Golaba-Behmela (SGB) | Gigantyzm, wady rozwojowe, guzy zarodkowe | GPC3(Xq26) | XR | Wysokie u chłopców, bliżej nieokreślone |
| HPT-JT (zespół guzów przytarczyc i guzów szczęki) | Gruczolaki rzadziej raki przytarczyc w młodym wieku, włóknisto-kostne guzy szczęki, guz Wilmsa | HRPT2(1q21-q31) | AD | Wysokie, bliżej nieokreślone |
| Perlmana | Macrosomia, visceromegalia, dysmorfie twarzy, wnętrostwo | ? | AR | <25% wśród rodzeństwa |
| Trisomia18 | Liczne wady wielu narządów, upośledzenie umysłowe, guz Wilmsa | | | Niskie, bliżej nieokreślone |
| Izolowany przerost połowiczy | Połowiczy przerost ciała | ? | ? | <5% |
| Aniridia | Aniridia | PAX6 | ? | 1,5% |
| Raka sutka-jajnika | Raki sutka i jajnika | BRCA1 (17q11), BRCAX | AD | Niskie |
| Li-Fraumeni | Sarcoma, białaczki, guzy mózgu, raki sutka | p53 (17p13) | AD | Niskie |
| Neurofibromatosis-typ1 | Nerwiakowłókniaki, plamy typu ""café-au-lait"", guzki Lischa, glejaki nerwu wzrokowego" | NF1 (17q11) | AD | Niskie |
| Blooma | "Niski wzrost, ""erythemata"" skóry, białaczki, chłoniaki, raki jelita grubego i inne nowotwory" | BLM (15q26) | | Niskie |
AD – autosomalne dominujące, AR – autosomalne recesywne, XR – recesywne sprzężone z chromosomem X
Sposób dziedziczenia odpowiada dziedziczeniu autosomalnemu dominującemu z niepełną penetracją.
W przypadkach nephroblastoma na bazie powyższych mutacji guzy występują częściej obustronnie niż w przypadkach bez uchwytnych predyspozycji genetycznych do nowotworów (ok. 20% versus ok. 5%) (1).
Zasady diagnostyki genetycznych uwarunkowań guza Wilmsa
Genetyczne uwarunkowania guzów Wilmsa rozpoznawane są rzadko, co wynika m.in. z tego, że nie jest doceniane znaczenie:
1) zbierania wśród dalszych krewnych danych rodowodowych niezbędnych dla rozpoznania rodzinnych postaci nephroblastoma;
2) kojarzenia występowania nephroblastoma z silną agregacją innych nowotworów np. raka sutka i jajnika lub nerwiaków, mięsaków w rodzinach z mutacją NF1;
3) pełnej oceny zmian dysmorficznych pozwalającej na wykrycie genetycznych uwarunkowań guza Wilmsa, którym towarzyszą wady rozwojowe.
Podobnie jak w innych zespołach dziedzicznej predyspozycji do nowotworów, w rozpoznawaniu guza Wilmsa należy uwzględnić następujące etapy:
– wnikliwą ocenę danych rodowodowo-klinicznych:
a) występowanie guza Wilmsa nie tylko wśród krewnych I° i II°, ale także wśród krewnych III° i IV°;
b) występowanie w rodzinie agregacji innych nowotworów;
– badanie przedmiotowe;
– badanie cytogenetyczne;
– badanie molekularne DNA (4).
Badanie cytogenetyczne jest przydatne szczególnie w diagnostyce dziedzicznych postaci guza Wilmsa powstałych de novo. Badanie to jest stosunkowo tanie i bardziej dostępne niż badanie molekularne. Wykazano, że ocena kariotypu jest efektywna w ujawnianiu przemieszczenia w obrębie locus WT2 u pacjentów z BWS, natomiast hybrydyzacja in situ jest czułą metodą w ocenie występowania dużych delecji WT1 u osób z zespołem WAGR.
W pracowniach molekularnych wykonujących badania naukowe możliwa jest analiza pełnej sekwencji genów WT1, p53, GPC3, dzięki której można wykryć obecność mutacji punktowych. Niestety pozostałe geny odpowiedzialne za powstawanie dziedzicznego nephroblastoma (WT2, FWT1, FWT2) nie zostały dotąd sklonowane.
Badania kontrolne w rodzinach z wysokim ryzykiem guza Wilmsa
W rodzinach z predyspozycją do guza Wilmsa należy stosować USG jamy brzusznej co 3 miesiące od urodzenia do 8 roku życia, a następnie co 6 miesięcy do 12 roku życia (po 12 roku życia rzadziej) (1, 5). W przypadkach wykrycia w USG zmian o niejasnym charakterze lub stwierdzenia pozostałości zarodkowej tkanki nefrogennej (istnieje pogląd, że nephroblastoma wywodzi się z tkanki nefrogennej) zaleca się wykonanie rezonansu magnetycznego lub tomografii komputerowej (1, 6). Ponadto w zespole Beckwith-Wiedemanna (BWS), z powodu zwiększonego ryzyka innych nowotworów, w pierwszych latach życia schemat badań powinien być uzupełniony o oznaczanie α-fetoproteiny w celu wykrycia ewentualnego hepatoblastoma.Niektórzy autorzy zalecają również wykonanie okresowych badań rtg klatki piersiowej i oceny wydalania VMA (kwasu wanilino-migdałowego) w celu wczesnego wykrycia neuroblastoma (1).
RAK NERKI (RCC)
Rak nerki stanowi ok. 3% nowotworów złośliwych osób dorosłych. W Polsce rozpoznawanych jest corocznie ponad 2000 nowych przypadków (7). Spośród wszystkich RCC najczęściej występującym jest rak jasnokomórkowy (CCRC) – stanowi on ok. 80% wszystkich rozpoznawanych raków nerki (7). Częstość występowania RCC związanego z wysokim genetycznym ryzykiem wielu autorów określa na 1-2% wszystkich RCC (8).
Dotychczas opisano 19 zespołów dziedzicznej predyspozycji do nowotworów, w przebiegu których w nerkach mogą powstać: CCRC, rak brodawkowaty (PRCC) lub rak z nabłonka przejściowego.
Rak jasnokomórkowy (CCRC)
Najlepiej poznanym zespołem genetycznej predyspozycji do nowotworów, w przebiegu którego może rozwinąć się CCRC jest zespół VHL opisany w innym rozdziale niniejszego opracowania (7, 9, 10, 11, 12).
Najczęściej jednak rozpoznawanym zespołem jest rodzinny rak jasnokomórkowy nerki (F-CCRC) specyficzny narządowo, w przebiegu którego stwierdza się co najmniej 2 przypadki CCRC, a brak cech zespołu VHL (8, 13, 14, 15).
Według naszych danych wśród wszystkich CCRC 5% stanowią rodziny spełniające kryteria definitywne F-CCRC zaś 13% stanowią przypadki z rodzin z pojedynczym zachorowaniem na CCRC jednak z wysokim prawdopodobieństwem rozwoju kolejnych CCRC.
Dziedziczną predyspozycję do nowotworów najbardziej precyzyjnie można rozpoznać, gdy stwierdzi się mutację konstytucyjną w jednym z genów odpowiedzialnych za ich powstawanie. Dzięki możliwości oceny mutacji w genach BRCA1, BRCA2, MSH2, czy MLH1niezwykłą skuteczność diagnozowania osiągnięto w przypadku wysokiej predyspozycji do raków piersi, jajnika, jelita grubego i trzonu macicy.
W przypadku F-CCRC nie znaleziono dotychczas genu odpowiedzialnego za jego powstawanie. W diagnostyce niektórych rodzin z F-CCRC przydatne jest badanie cytogenetyczne.
W dotychczasowej literaturze opisano 3 rodziny z F-CCRC, w których stwierdzono związek powstawania CCRC z występowaniem konstytucyjnych translokacji. W 1979 roku Cohen i jego współpracownicy opisali rodzinną translokację zrównoważoną pomiędzy chromosomami 3 i 8 [t(3;8)(p14,2;q24,1)] będącą przyczyną powstawania obustronnych CCRC w młodym wieku – w przedziale wiekowym 37 r.ż.-59 r.ż. (16). U jednej nosicielki tej translokacji wystąpił wieloogniskowy brodawkowaty i anaplastyczny rak tarczycy. Gemmill i współpracownicy w 1998 roku wysnuli teorię, że t(3;8) prowadzi do fuzji genów FHIT i TRC8, z których być może powstaje gen powodujący zwiększone ryzyko wystąpienia raka nerki. W 1988 roku Kovacs opisał rodzinę z konstytucyjną translokacją t(3;6)(p13;q25,1). U nosiciela tej translokacji w 53 roku życia wystąpił obustronny, wieloogniskowy rak jasnokomórkowy nerki (17, 18).
Kolejny odnotowany w piśmiennictwie przypadek rodziny z rodzinnym rakiem nerki, którego przyczyną była translokacja został opisany w 1998 roku przez zespół Koolen i Bodmera (19, 20). W rodzinie tej u 4 nosicieli translokacji zrównoważonej t(2;3)(q35;q21) wystąpiły raki nerki w wieku 40, 53, 54, 68 lat, w tym 3 raki jasnokomórkowe. Ponadto u jednego z nosicieli rozpoznano raka płaskonabłonkowego pęcherza moczowego.
Taką samą translokację stwierdzono w 1996 roku w naszym Ośrodku u dwóch braci z obustronnym rakiem jasnokomórkowym nerki, u których w rodzinie 5 dalszych osób zmarło również z powodu raka nerki (21).
Van Kessel ze współpracownikami przebadali 57 nosicieli różnych translokacji zrównoważonych chromosomu 3 z 10 rodzin. U czterech badanych stwierdzono raka nerki. Na podstawie swych obserwacji wyciągnęli wniosek, że u nosicieli translokacji chromosomu 3 jest zwiększone ryzyko raka nerki, szczególnie, jeśli miejsce złamania znajduje się blisko centromeru (22). Raki te występują w młodym wieku (ok. 45 lat), są wieloogniskowe i obustronne.
W większości przypadków F-CCRC opisanych w literaturze nie znaleziono mutacji będących przyczyna agregacji CCRC w rodzinach (23, 24, 25, 26). Ostatnio w naszym Ośrodku wykazano, że istotną przyczyną powstawania raka jasnokomórkowego nerki jest konstytucyjna zmiana I157T w genie CHEK2 (27).
Nadal jednak podstawową metodą rozpoznawania F-CCRC jest ocena danych rodowodowo-klinicznych.
Zdefiniowane przez nasz zespół kryteria rodowodowo – kliniczne oparte są o ocenę krewnych I° chorego z CCRC („nuclear pedigree” kryteria), pozwalające na rozpoznawanie z dużym prawdopodobieństwem rodzin z F-CCRC pomimo tego, że wśród najbliższych krewnych (tj. krewnych I°) nie stwierdzono przypadków CCRC.
Na podstawie naszych analiz stwierdziliśmy, że w rodzinach z pojedynczym CCRC można wysunąć podejrzenie F-CCRC stosując jako kryterium (ryc. 1):
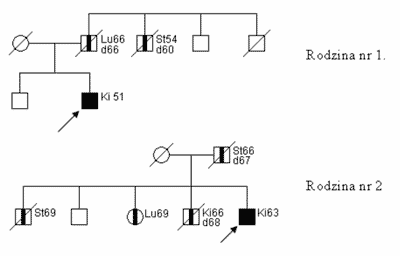
Ryc.1. Rodowody rodzin z F-CCRC.
– zdiagnozowanie CCRC poniżej 55 roku życia
lub
– wystąpienie raka żołądka lub raka płuca u krewnych I° pacjenta z CCRC.
Badania kontrolne w rodzinach z F-CCRC
Cechą charakterystyczną CCRC jest bezobjawowy początek choroby. Dolegliwości kliniczne pojawiają się dopiero w stadium znacznego zaawansowania nowotworu. W świetle danych literaturowych wydaje się, że właściwe postępowanie lekarskie w rodzinach z F-CCRC rzeczywiście może zwiększać szansę wczesnego wykrywania nowotworu, a w związku z tym skutecznego leczenia (25, 28, 29).
Jak dotąd brak jednakże zweryfikowanych programów badań kontrolnych wykrywania wczesnych F-CCRC.
Levinson sugeruje, że członkowie rodzin z F-CCRC powinni mieć wykonywane jedynie USG nerek, co 2-3 lata począwszy od 30 roku życia (22). Ten schemat akceptują również inni autorzy (2, 30). Jest to jednak program badań przyjęty arbitralnie.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Cybulski C, et al.: Nowotwory dziedziczne u dzieci – guz Wilmsa. Wsp Onk 2002, 6: 300-7.
2. Dome JS, Coppes MJ: Recent advances in Wilms tumor genetics. Current opinion in pediatrics 2002, 14: 5-11.
3. Ruteshouser EC, Huff V: Familial Wilms Tumor. Am J Med Genet 2004, 129: 29-34.
4. Zajączek S, Lubiński J: Zasady poradnictwa genetycznego u rodzin o podwyższonym ryzyku choroby nowotworowej. Nowotwory 1999, 49: 71-2.
5. Little M, Wells C: A clinical overview of WT1 gene mutations. Human Mutat 1997, 9: 209-25.
6. Borer JG, et al.: Renal findings on radiological followup of patients with Beckwith-Wiedemann syndrome. J Urol 1999, 161: 235-9.
7. Borkowski A, Czaplicki M: Nowotwory i torbiele nerek. Wyd 1. PZWL, 2002.
8. Goldman SM, et al.: Renal cell carcinoma diagnosed in three generations of a single family. South Med J 1979, 72: 1457-9.
9. Krzystolik K i wsp.: Wczesna diagnostyka bezobjawowych raków nerek w rodzinach z zespołem von Hippel-Lindau w Polsce. Urol Pol 1998, 51: 171-81.
10. Neumann HP: Basic criteria for clinical diagnosis and genetic counseling in von Hippel-Lindau syndrome. J Vasc Dis 1987, 16: 220-6.
11. Neumann HPH, et al.: Prevalence, morphology and biology of renal cell carcinoma in von Hippel-Lindau disease compared to sporadic renal cell carcinoma. J Urol 1998, 160: 1248-54.
12. Neumann HPH, Zbar B: Renal cysts, renal cancer and von Hippel-Lindau disease. Kidney Int 1997, 51: 16-26.
13. Erlandsson R, et al.: Do human renal cell carcinomas arise by a double-loss mechanism? Cancer Genet Cytogenet 1988, 36: 197-202.
14. Franksson C, et al.: Renal carcinoma (hypernephroma) occurring in 5 siblings. J Urol 1972, 108: 58-61.
15. Reddy ER: Bilateral renal cell carcinoma – unusual occurrence in three members of one family. Br J Radiol 1981, 54: 8-11.
16. Cohen AJ, et al.: Hereditary renal-cell carcinoma associated with a chromosomal translocation. N Engl J Med 1979, 301: 592-5.
17. van Kessel G, et al.: Renal cell cancer: chromosome 3 translocations as risk factor. J Nat Cancer Inst 1999, 91: 1159-60.
18. Maher ER, Yates JRW: Familial renal cell carcinoma: clinical and molecular genetic aspects. Br J Cancer 1991, 63: 176-9.
19. Bodmer D, et al.: An alternative route for multistep tumorigenesis in a novel case of hereditary renal cell cancer and t(2:3)(q35;q21) chromosome translocation. Am J Hum Genet 1998, 62: 1475-83.
20. Koolen MI, et al.: A familial case of renal cell carcinoma and a t(2;3) chromosome translocation. Kidney Int 1998, 53: 273-5.
21. Borówka A, Zajączek S: Rodzinne występowanie raka jasnokomórkowego nerki. Doniesienie zjazdowe: 26 Kongres PTU, Poznań 1996.
22. Kovacs G, Brusa P, De Rirse W: Tissue-specific expression of a constitutional 3;6 translocation: development of multiple bilateral renal-cell carcinomas. Int J Cancer 1989, 43: 422-7.
23. Levinson AK, et al.: Familial renal carcinoma: hereditary or coincidental? J Urol 1990, 144: 849-51.
24. Li FP, Marchetto DJ, Brown RS: Familial renal carcinoma. Cancer Genet Cytogenet 1982, 7: 271-5.
25. Teh B, Giraud S, Sari F: Familial non- VHL non-papillary clear-cell renal cancer. Lancet 1997, 349: 848-9.
26. Woodward ER, et al.: Familial clear cell renal carcinoma (FCRC): clinical features and mutation analysis of the VHL, MET, and CUL2candidate genes. J Med Genet 2000, 37: 348-53.
27. Cybulski C, et al.: CHEK2is the multiorgan cancer susceptibility gene. Am J Hum Genet 2004, 75: 1131-5.
28. Herring JC, et al.: Parenchymal sparing surgery in patients with hereditary renal cell carcinoma: a 10-years experience. J Urol 2001, 165: 777-81.
29. Schmidt L, et al.: Two North American families with hereditary papillary renal carcinoma and identical novel mutations in the MET proto-oncogene. Cancer Res 1998, 58: 1719-22.
30. Walther MM, et al.: Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol 1999, 161: 1475-9.
31. Bosniak MA, Rofsky NM: Problems in the detection and characterization of small renal masses. Radiology 1996, 198: 638-41.
32. Choyke PL, et al.: Linehan WM. von Hippel-lindau disease: radiologic screening for visceral manifestation. Radiology 1990, 174: 815-20.
33. Curry NS: Small renal masses (lesions smaller than 3 cm): imaging evaluation and management. Am J Radiol 1995, 164: 355-62.
34. Jamis Dow CA, et al.: Small (
35. Lightfoot N, et al.: Impact of noninvasive imaging on increased incidental detection of renal cell carcinoma. Eur Urol 2000, 37: 521-7.
36. Shinohara N, et al.: Nephron sparing surgery for renal cell carcinoma in VHL disease. J Urol 1995, 154: 2016-9.
37. Walther MM, Linehan WM: Nephron sparing surgery for renal cell carcinoma in von Hippel-Lindau disease. J Urol 1996, 156: 480-1.
38. Zbar B, Lerman M: Inherited carcinomas of the kidney. Adv Cancer Res 1998, 75: 163-201.
39. Zbar B, et al.: Hereditary papillary renal cell carcinoma: clinical studies in 10 families. J Urol 1995, 53: 907-12.
40. Zbar B, et al.: Hereditary papillary renal cell carcinoma. J Urol 1994, 151: 561-6.
41. Schmidt L, et al.: Germline and somatic mutations in tyrosine kinase domain of MET proto-oncogene in papillary renal carcinomas. Nat Gen 1997, 16: 68-73.
otrzymano: 2008-04-23
zaakceptowano do druku: 2008-07-04
Adres do korespondencji:
*Aleksandra Tołoczko-Grabarek
Międzynarodowe Centrum Nowotworów Dziedzicznych,
Zakład Genetyki i Patomorfologii,
Pomorska Akademia Medyczna
ul. Połabska 4, 70-115 Szczecin
tel.: (0-91) 466 -15-32
e-mail: otjg@poczta.onet.pl
Postępy Nauk Medycznych 8/2008Strona internetowa
czasopisma Postępy Nauk MedycznychPozostałe artykuły z numeru 8/2008: