© Borgis - Medycyna Rodzinna 3/2010, s. 11-15
*Sergiusz Jóźwiak1, Monika Dudzisz-Śledź2
Stwardnienie guzowate – możliwości diagnostyczne i terapeutyczne
Tuberous sclerosis complex – diagnostic and therapeutic possibilities
1Klinika Neurologii i Epileptologii Instytutu „Pomnik Zdrowia Dziecka” w Warszawie
Kierownik Kliniki: prof. dr hab. n. med. Sergiusz Jóźwiak
2Novartis Poland Sp. z o.o., Warszawa
Kierownik: dyrektor medyczny dr n. med. Jan Krzysztof Żelazowski
Summary
Tuberous sclerosis complex (TSC) is one of the most common neurocutaneous diseases. The prevalence of TSC is 1:6000 in general population. The disease belongs to neuroectodermal dysplasias known as phacomatoses. TSC is the result of genes TSC1 and TSC2 mutations. The disease is characterized by diverse clinical phenotype and prolonged and progressive course. The most common clinical features of this disease include epilepsy and developmental delay. It should be emphasized, that mental development is normal in almost 50% patients. Genetically determined potential risk of developing tuberous lesions, hamartomas or benign tumors of different organs: heart, kidney, brain, liver, skin is characteristic for this condition. Signs and symptoms appear in different periods of development, some of them could appear in the prenatal life. The diagnosis of TSC is based on precisely defined clinical criteria and imaging studies. The patients should be closely controlled with imaging studies due to increased risk of tumors development. The treatment of TSC is primarily symptomatic, comprehensive and based on involved organs. It should be conducted in specialized centers. Especially problematic is epilepsy management. Epilepsy is often drug-resistant and needs to be treated with combined therapy. The treatment should be conducted under supervision of experienced neurologist. Recently big progress has been made regarding TSC diagnosis, monitoring and treatment, leading to significant improvement of care of patients with TSC.

Wstęp
Stwardnienie guzowate (TSC, ang. tuberous sclerosis complex) in. choroba Bourneville'a należy do grupy chorób określanych wspólnym mianem fakomatozy lub dysplazje neuroektodermalne, które charakteryzują się występowaniem zaburzeń rozwojowych w obrębie trzech listków zarodkowych, a zwłaszcza ektodermy. Częstość występowania choroby szacowana jest na 1: 6 000 (1). Na całym świecie na tę chorobę choruje 1,5 miliona ludzi (2). Choroba odznacza się zróżnicowanym obrazem klinicznym, a do jej charakterystycznych objawów należą: padaczka, opóźnienie rozwoju, zmiany skórne oraz zmiany rozrostowe w układzie nerwowym, narządzie wzroku, nerkach, sercu i wątrobie. Choroba ma charakter przewlekły i postępujący (1, 3).
Choroba jest dziedziczona jako cecha autosomalna dominująca z wysoką penetracją i zmienną ekspresją genu, co jest powodem dużej różnorodności obrazu klinicznego. Za jej rozwój odpowiedzialne są mutacje dwóch genów: TSC1 (9q34) i TSC2 (16p13), przy czym mutacje genu TSC2 związane są z cięższym przebiegiem choroby (1, 4, 5, 6). Mutacje w obrębie genów TSC1 i TSC2 stwierdzane są u około 85% pacjentów spełniających kliniczne kryteria rozpoznania TSC. U około 15% pacjentów z TSC nie stwierdza się jednak żadnej mutacji w obrębie genów TSC1 i TSC2, a rozpoznanie stawiane jest na podstawie obrazu klinicznego. Przyczyną tego faktu może być mozaikowatość gonadalna (zmutowane komórki są tylko w obrębie gonad i u pozornie zdrowej osoby nie stwierdza się cech stwardnienia guzowatego) lub też udział innych mechanizmów w patogenezie TSC. Nie wiadomo również, dlaczego te same mutacje u różnych osób warunkują odmienny obraz i przebieg kliniczny choroby, a więc osoby o tym samym genotypie mogą mieć różne cechy fenotypowe (7, 8). W około 60% (9) – 70% (8) przypadków stwierdza się nowe mutacje. Objawy chorobowe pojawiają się wraz ze wzrostem dziecka, a u dzieci w pierwszym roku życia rozpoznanie choroby może być szczególnie trudne (8). Geny TSC1 i TSC2 należą do genów supresorowych nowotworów, czyli hamujących rozwój nowotworów. Białka kodowane przez te geny, hamartyna i tuberyna, odgrywają istotną rolę w hamowaniu szlaku przekazywania sygnałów przebiegającego z udziałem kinazy mTOR (ang. mammalian target of rapamycin) (8). Aby doszło do wystąpienia choroby, konieczna jest inaktywacja obu prawidłowych alleli genu TSC1 lub TSC2 (10).
W ciągu ostatnich dwudziestu lat dokonał się olbrzymi postęp w zakresie diagnostyki, monitorowania przebiegu i leczenia stwardnienia guzowatego, co doprowadziło do istotnej poprawy opieki nad pacjentami z TSC (4, 7). Na podstawie licznych badań przedklinicznych ustalono, iż hamartyna i tuberyna pełnią funkcje regulatorowe szlaków przekazywania sygnałów Rheb i mTOR. Odkrycia te pozwoliły na rozpoczęcie dalszych prac nad molekularnymi mechanizmami choroby oraz nad możliwościami ich leczenia z uwzględnieniem inhibitorów kinazy mTOR (ang. mammalian target of rapamycin) (7, 11).
Stwardnienie guzowate jest wyjątkowo skomplikowaną chorobą, dotykającą różnych układów narządów w różnym stopniu i w zróżnicowany sposób, w różnym okresie życia pacjentów (4).
Obraz kliniczny
Objawy kliniczne, które wskazują na rozpoznanie TSC są bardzo różnorodne i w dużej mierze uzależnione są od wieku pacjenta. U dzieci objawami, które naprowadzają lekarza na to rozpoznanie są najczęściej guzki ( rhabdomyosarcoma) serca lub drgawki, u dorosłych są to zwykle zmiany skórne, nerkowe lub płucne. Często choroba rozpoznawana jest dopiero u pacjenta w wieku dorosłym, po wystąpieniu drgawek lub krwotoku z guza nerki (4).
Najczęstsze zmiany w TSC dotyczą OUN i skóry. Mózg i skóra zajęte są u 90-95% pacjentów z TSC (4, 7). Większość zmian w przebiegu TSC uwidacznia się u dzieci powyżej 3. roku życia, co utrudnia ustalenie rozpoznania we wcześniejszym wieku (7).
Do głównych zmian skórnych zalicza się: znamiona bezbarwne (ryc. 1), mnogie guzkowe naczyniakowłókniaki na twarzy (ryc. 2), płaskie włókniaki w okolicy czołowej, zmiany na skórze określane jako „skóra szagrynowa”, czyli mnogie zmiany o średnicy 0,5-10 cm zwykle barwy cielistobrązowej przypominające włókniaki (ryc. 3), rozsiane na tułowiu szczególnie w okolicy lędźwiowo-krzyżowej, włókniaki okołopaznokciowe, uszypułowane włókniaki okolicy karkowej i szyjnej, zmiany typu „confetti” czyli liczne plamiste drobne odbarwienia o średnicy 1-3 mm zwykle w okolicy przedramion i podudzi (12, 13).

Ryc. 1. Plama odbarwieniowa na skórze w przebiegu TSC.
Ryc. 2. Naczyniakowłókniaki na twarzy u mężczyzny z TSC.
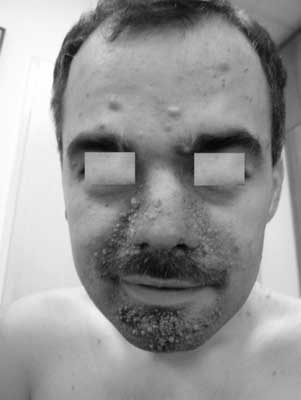 Ryc. 2. Naczyniakowłókniaki na twarzy u mężczyzny z TSC.
Ryc. 2. Naczyniakowłókniaki na twarzy u mężczyzny z TSC.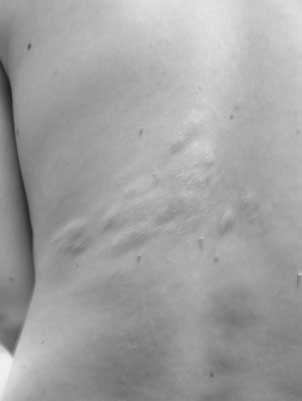 Ryc. 3. Skóra szagrynowa u pacjenta z TSC.
Ryc. 3. Skóra szagrynowa u pacjenta z TSC.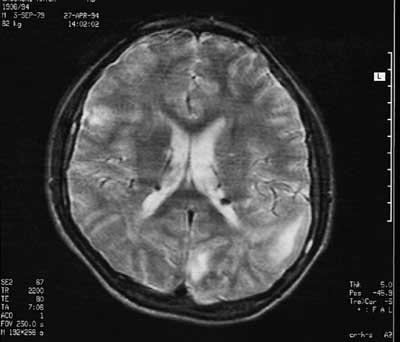 Ryc. 4. Guzki okołokomorowe i guzki korowe widoczne w obrazie MRI pacjenta z TSC.
Ryc. 4. Guzki okołokomorowe i guzki korowe widoczne w obrazie MRI pacjenta z TSC.