© Borgis - Anestezjologia Intensywna Terapia 3/2007, s. 166-174
*Piotr Smuszkiewicz1, Edyta Szałek2, Hanna Tomczak3, Iwona Trojanowska1, Maciej Błaszyk1
Farmakokinetyczno-farmakodynamiczne zasady stosowania antybiotyków u chorych leczonych z powodu sepsy
Pharmacokinetic and pharmacodynamic considerations of antimicrobial therapy in sepsis
1Klinika Anestezjologii, Intensywnej Terapii i Leczenia Bólu AM w Poznaniu
kierownik: prof. dr hab. n. med. L. Drobnik
2Zakład Farmacji Klinicznej i Biofarmacji AM w Poznaniu
kierownik: prof. dr hab. n. farm. E. Grze?kowiak
3Centralne Laboratorium Mikrobiologiczne Pracownia Bakteriologii SPSK Nr 2 w Poznaniu
kierownik: dr n. farm. H. Tomczak
Summary
Contemporary antimicrobial therapy should be directed by objective criteria which allow for maximal effectiveness of treatment with minimal side effects. ITU patients are at special risk because of profound changes in the organism that may alter both the pharmacokinetics and the pharmacodynamics of drugs. Therapeutic concentrations are frequently not achieved, leading to multiple bacterial resistance. Typical parameters (Cl, Vd, T0,5, AUC, Cmax) do not adequately describe the altered PK/PD in sepsis. Three other indicators, Cmax/MIC, AUC/MIC, and T>MIC allow for better prediction of therapeutic effects. For drugs where effectiveness is concentration dependent (eg. aminoglycosides), the Cmax/MIC indicator should be used. For the successful use of the AUC dependent drugs (fluorchinolones), the AUC/MIC indicator should used and kept above>125. The T>MIC indicator is important for betalactam antibiotics, macrolides and clindamycin.
The standardization of these indicators should improve the effectiveness of antimicrobial therapy is sepsis.

Zasady leczenia chorego w stanie septycznym oparte sš na terapii podtrzymujšcej funkcje narzšdów, leczeniu chirurgicznym oraz stosowaniu antybiotyków. Antybiotyki sš najczę?ciej przepisywanš grupš leków chorym w stanach krytycznych, stšd też konieczno?ć dokładnego okre?lenia zasad ich stosowania.
Celem antybiotykoterapii jest likwidacja mikroorganizmów w miejscu zakażenia. Uzyskuje się to poprzez osišgnięcie odpowiedniego, efektywnego stężenia antybiotyku przy równoczesnym unikaniu działań toksycznych. Stężenie skuteczne to takie, które utrzymuje się powyżej minimalnego stężenia hamujšcego (MIC – Minimal Inhibitory Concentration) lub minimalnego stężenia bakteriobójczego (MBC – Minimal Bactericidal Concentration).
Odmienno?ć stosowania antybiotyków u chorych z ciężkimi zakażeniami, hospitalizowanych w oddziałach intensywnej terapii, w porównaniu z antybiotykoterapiš innych zakażeń wynika z towarzyszšcych zaburzeń hemodynamicznych i metabolicznych (wzrost przepuszczalno?ci ?ródbłonków naczyniowych, hipoalbuminemia, zwiększenie objęto?ci wody pozanaczyniowej oraz rzutu serca), które w zasadniczy sposób zmieniajš parametry farmakokinetyczne i farmakodynamiczne antybiotyków. Nieuwzględnianie tych czynników i wynikajšce z tego niewła?ciwe dawkowanie antybiotyków, przyczyniajš się do niepowodzeń terapeutycznych lub opó?nionej odpowiedzi klinicznej. Prowadzi to równocze?nie do niepotrzebnej ekspozycji na antybiotyk, promujšc kolonizację i rozwój szczepów wieloopornych [1]. Niekiedy, pomimo wła?ciwego stosowania antybiotyków u chorych w ciężkiej sepsie, obserwuje się nieskuteczno?ć terapii z powodu upo?ledzonej funkcji układu immunologicznego oraz dostępno?ci antybiotyku w odpowiednim stężeniu w miejscu zakażenia (zła penetracja wynikajšca z wła?ciwo?ci antybiotyku, zjadliwo?ć mikroorganizmów, zagęszczenie patogenów w miejscu zakażenia – inoculum) [2].
Terminologia i charakterystyka pk/pd antybiotyków
Aby okre?lić optymalnš dawkę, czas i sposób podawania antybiotyków, a tym samym poprawić skuteczno?ć terapii, należy mieć na uwadze wła?ciwo?ci farmakokinetyczne (PK) i farmakodynamiczne (PD) poszczególnych grup antybiotyków [3]. Rekomendacje te wynikajš z zależno?ci pomiędzy wrażliwo?ciš czynnika etiologicznego in vitro (MIC) a wska?nikami PK/PD in vivo.Taka strategia pomaga w zapobieganiu rozwojowi kolonizacji i szerzeniu się wielooporno?ci.
PK antybiotyków odnosi się do ich rozmieszczenia w organizmie oraz opisuje profil zależno?ci stężenia leku od czasu, podczas gdy PD zwišzane jest ze stężeniem i działaniem przeciwdrobnoustrojowym antybiotyku w jego miejscu docelowym. Posługujšc się koncepcjš PK/PD należy pamiętać, że tylko niezwišzana z białkami frakcja leku jest formš aktywnš pod względem mikrobiologicznym i zdolnš do przechodzenia przez ?ródbłonek do przestrzeni pozanaczyniowej PK opisuje przebieg procesów składajšcych się na losy leku i/lub jego metabolitów w organizmie: absorpcję i dyspozycję. Przez dyspozycję rozumie się dystrybucję i eliminację (biotransformację i wydalanie) leku.
Zależno?ci między poszczególnymi etapami okre?lajšcymi losy leku w ustroju sš ilo?ciowo wyrażane poprzez odpowiednie parametry farmakokinetyczne, z których najistotniejszymi sš: objęto?ć dystrybucji Vd, klirens leku Cl, biologiczny okres półtrwania t0,5, stężenie maksymalne Cmax i minimalne Cmin leku we krwi oraz dostępno?ć biologiczna F. Podaż antybiotyków w OIT zwykle odbywa się drogš dożylnš, a to oznacza 100% wchłanianie leku (F=1). Dystrybucja leku pomiędzy kompartmentami uzależniona jest od takich zmiennych jak: rzut minutowy serca, regionalny przepływ krwi, całkowita zawarto?ć wody w przestrzeni wewnštrz- i pozakomórkowej oraz zasoby rezerw fizjologicznych tych narzšdów, które odpowiedzialne sš za metabolizm i eliminację leków. Dystrybucja antybiotyków zależy również od ich fizykochemicznych wła?ciwo?ci tj. rozpuszczalno?ci w wodzie, w tłuszczach, wišzania z białkami osocza oraz procesów metabolizmu i wydalania.
Antybiotyki, ze względu na rozpuszczalno?ć można podzielić na hydrofilne i lipofilne. Do pierwszej grupy zalicza się: b-laktamy (penicyliny, cefalosporyny, karbapenemy, monobaktamy), glikopeptydy i aminoglikozydy. Antybiotyki te charakteryzujš się ograniczonš objęto?ciš dystrybucji i eliminacjš drogš nerkowš w niezmienionej postaci, natomiast farmakodynamicznie – utrudnionym przechodzeniem przez błony biologiczne i w zwišzku z tym brakiem aktywno?ci w zakażeniach wewnštrzkomórkowych np. Legionella, Chlamydia.Antybiotyki lipofilne (fluorochinolony, makrolidy, tetracykliny, chloramfenikol, rifampicyna) przeciwnie, majš dużš objęto?ć dystrybucji, podlegajš metabolizmowi oraz łatwo dyfundujš przez błony i bariery anatomiczne (np. bariera krew-mózg), przez co wykazujš dużš skuteczno?ć przeciwko patogenom wewnštrzkomórkowym (tab. I) [1]. W obu grupach antybiotyków występujš jednak nieliczne wyjštki.
Tab. I. Klasyfikacja antybiotyków wg wła?ciwo?ci PK/PD [wg 1]
| Hydrofilne | Lipofilne |
b-laktamy
Glikopeptydy
Aminoglikozydy | Ograniczona objęto?ć dystrybucji.
Nie dyfundujš przez błony plazmatyczne.
Nieaktywne wobec patogenów ?ródkomórkowych.
Eliminacja w postaci niezmienionej drogš nerkowš. | Makrolidy
Fluorochinolony
Tetracykliny
Rifampicyna
Klindamycyna | Duża objęto?ć dystrybucji.
Łatwa dyfuzja przez błony plazmatyczne.
Aktywne wobec patogenów ?ródkomórkowych.
Eliminacja głównie przez metabolizm wštrobowy. |
Po podaniu dożylnym osišgane jest wysokie stężenie szczytowe antybiotyku we krwi, po czym następuje jego dystrybucja do przedziału pozanaczyniowego i komórkowego. Różne systemy transportu biorš udział w przechodzeniu antybiotyków do komórek. Transport ten odbywa się: w mechanizmie endocytozy (aminoglikozydy – lecz ich aktywno?ć ?ródkomórkowa jest ograniczona), prostej dyfuzji (fluorochinolony), lub w mechanizmie aktywnego transportu (makrolidy i klindamycyna). W wielu przypadkach jednakże stopień penetracji do przedziału komórkowego niekoniecznie koreluje z aktywno?ciš mikrobiologicznš, prawdopodobnie ze względu na niekorzystne rozmieszczenie antybiotyku w samej komórce [4]. Najsilniejszy efekt w leczeniu infekcji wewnštrzkomórkowych wywierajš makrolidy oraz fluorowane chinolony.
W przypadku intensywnej terapii szczególne znaczenie ma stężenie antybiotyków w wydzielinie oskrzelowej, plwocinie i mišższu płuc. Stężenie osišgane w plwocinie przez większo?ć antybiotyków jest niskie, podczas gdy w mišższu płuc stosunkowo wysokie. Antybiotyki takie jak ciprofloksacyna, cefotaksym i erytromycyna uzyskujš znaczšco wyższe stężenie w płucach aniżeli w osoczu i wydajš się wła?ciwymi lekami przy zwalczaniu infekcji płucnych [4, 5].
Patofizjologia sepsy a farmakokinetyka antybiotyków
Znalezienie wła?ciwej zależno?ci pomiędzy chorym, patogenem i chemioterapeutykami może być trudne u osób w ciężkim stanie ogólnym. Schematy dawkowania antybiotyków pochodzš bowiem głównie z badań z udziałem zdrowych ochotników i chorych, u których nie występuje dysfunkcja wielonarzšdowa.
Odpowied? zapalna zwišzana z sepsš wywołuje szereg powszechnie znanych zaburzeń ogólnoustrojowych. Towarzyszšcy zwykle sepsie podwyższony rzut serca i zwiększony przepływ nerkowy krwi poczštkowo powoduje zwiększenie klirensu kreatyniny, nasila filtrację kłębuszkowš i wydzielanie cewkowe eliminowanych przez nerki antybiotyków; jednak w miarę postępu choroby funkcje narzšdów (wštroba, nerki i serce) ulegajš upo?ledzeniu, co wydłuża t0,5 i zmniejsza Cl antybiotyków, a także sprzyja ich kumulacji [6]. Zaburzenia patofizjologiczne w ciężkich zakażeniach skutkujš dużym wzrostem Vd antybiotyków (tab. II) oraz sš przyczynš redukcji ich stężenia zarówno we krwi jak i płynie pozakomórkowym. Aby uzyskać wystarczajšce stężenie docelowe (CT) antybiotyku (głównie aminoglikozydów), rozsšdnym byłoby więc zwiększenie dawki (D), która jest wprost proporcjonalna do Vd [7]. Ponadto przy niezmienionym Cl zwiększona Vd antybiotyków zmniejsza proporcjonalnie wielko?ć stałej szybko?ci eliminacji (Ke), zgodnie ze wzorem: Cl = Vd x Ke. Skoro stała szybko?ci eliminacji zwišzana jest z t0,5 zależno?ciš: Ke = ln2/t0,5, to wzrost Vd wydłuża t0,5. Byłoby to korzystne dla antybiotyków, których aktywno?ć jest zależna od czasu działania [4], jednak stosujšc tzw. standardowe dawkowanie, może to powodować dłuższe utrzymywanie się stężeń subterapeutycznych, co skutkuje rozwojem wielooporno?ci. W tym scenariuszu, chcšc utrzymać efektywne stężenie, konieczne jest umiarkowane (Cmin/MIC 4-5) zwiększenie dawki antybiotyku (i często?ci podawania), aby zrekompensować zmianę Vd.
Tab. II. Parametry PK wybranych antybiotyków w ciężkiej sepsie
| Antybiotyk | Parametry |
| Vd (l kg-1) | t0,5 (h) |
Cefepim
Piperacylina/Tazobaktam
Imipenem
Meropenem
Ciprofloksacyna
Aminoglikozydy
Wankomycyna
Teikoplanina | 0,31
0,31
0,4
0,27
2
0,3
0,2-1,25
0,9-1,6 | 2,5
1,5
1,5
2,5
3,3
2-3
4-6
80-160 |
W ciężkiej sepsie redystrybucja kršżenia promuje przepływ krwi przez mózg i serce kosztem innych narzšdów. W badaniach na zwierzętach wykazano, że aminoglikozydy nie osišgajš wystarczajšcego stężenia w mikrokršżeniu we wczesnej fazie sepsy. Fakt występowania hipoperfuzji tkankowej i gorszej penetracji antybiotyków w tej fazie stał się implikacjš do stosowania wysokich dawek np. aminoglikozydów, aby uzyskać wysokie stężenie szczytowe [8].
Do zaburzeń patofizjologicznych dołšczajš czynniki jatrogenne zwiększajšce Vd. Agresywna resuscytacja płynowa w poczštkowej fazie sepsy oraz żywienie pozajelitowe zwiększajš przestrzeń pozanaczyniowš i przyczyniajš się do zmniejszenia stężenia antybiotyków (głównie hydrofilnych) [1]. Postuluje się, aby oprócz zwiększenia dawki, także mierzyć stężenie aminoglikozydów i wankomycyny u chorych otrzymujšcych znaczšce wsparcie płynowe [9]. Często niedocenianš przyczynš „utraty antybiotyków” (fałszywy wzrost Vd) jest obecno?ć drenów w jamie brzusznej i klatce piersiowej. Przechodzenie antybiotyków do płynu wysiękowego i znaczšcy objęto?ciowo drenaż istotnie przyczyniajš się do spadku ich stężenia [10]. Stosowanie ?rodków hemodynamicznie aktywnych także wišże się ze zmianš dawkowania antybiotyków. Aminy katecholowe czy leki moczopędne zwiększajš rzut minutowy serca, filtrację kłębuszkowš oraz nerkowy klirens antybiotyków [11].
Dysfunkcja narzšdowa oddziałujšc na mechanizmy eliminacji przedłuża biologiczny okres półtrwania leków i zmniejsza ich klirens, co czyni farmakokinetykę antybiotyków wysoce nieprzewidywalnš. Wykorzystywana w ciężkiej sepsie cišgła terapia nerkozastępcza zwiększa Cl antybiotyków i może prowadzić do ich adsorpcji na powierzchni filtra. Przy rzadko stosowanym rutynowym oznaczaniu stężeń antybiotyków we krwi, wszystkie wymienione czynniki powodujš trudno?ci w zróżnicowaniu niepowodzeń terapeutycznych, wynikajšcych bšd? to ze stężeń subterapeutycznych, bšd? z braku wrażliwo?ci patogenów. Tak więc, na różnych etapach zaawansowania ciężkiej sepsy zapotrzebowanie chorego na antybiotyki może być podobne, mniejsze lub większe w porównaniu z chorymi leczonymi z innych przyczyn.
Nie można też pomijać faktu, że – wyłšczajšc zakażenie krwi, gdzie patogeny sš zlokalizowane ?ródnaczyniowo – większo?ć infekcji dotyczy przedziału pozakomórkowego. Tymczasem odpowiednie stężenie antybiotyków powinno dotyczyć obu kompartmentów. Wykazano, co prawda, że stężenie osoczowe antybiotyków koresponduje ze stężeniem w płynie międzykomórkowym i równowaga pomiędzy tymi ?rodowiskami zwykle jest osišgana [12], jednakże u chorych z ciężkš sepsš może istnieć znaczna rozbieżno?ć pomiędzy tymi kompartmentami w zakresie stężenia niezwišzanej frakcji leku [13].
Spadek stężenia albumin wišżšcych leki o odczynie kwa?nym, występujšcy w sepsie potencjalnie zwiększa ilo?ć leku w postaci wolnej, podczas gdy obserwowany wzrost stężenia a-kwa?nej glikoproteiny, wišżšcej leki zasadowe, redukuje wolnš ilo?ć leku. Wydaje się, iż mimo spadku albumin i zwiększenia wolnej postaci leku, wzrost glikoprotein przebiega z większym nasileniem, co skutkuje zmniejszeniem stężenia antybiotyków w osoczu. Współistniejšcy często wzrost stężenia bilirubiny, wypierajšcej antybiotyki z połšczeń z albuminami, zwiększa wolnš frakcję leku oraz jego Vd. Dołšczajšca się zmienna aktywno?ć cytochromu P450 jest dopełnieniem obrazu zaburzeń PK/PD dla leków metabolizowanych. Dla większo?ci antybiotyków nie ma to większego znaczenia i nie wymaga istotnych modyfikacji dawkowania, niemniej, u chorych z poważnš dysfunkcjš wštroby należy rozważyć obniżenie dawek antybiotyków metabolizowanych w tym narzšdzie takich jak: chloramfenikol, klindamycyna, metronidazol, nafcylina, tetracyklina, cefotaksym, czy erytromycyna [4].
Większo?ć antybiotyków usuwana jest z organizmu w postaci niezmienionej drogš nerkowš, tak więc każda dysfunkcja (oliguria) powoduje ich kumulację. Duża grupa chorych poddawana jest w tym okresie cišgłej terapii nerkozastępczej, eliminujšcej antybiotyki z wielko?ciš odpowiadajšcš filtracji kłębuszkowej rzędu 35 ml min-1 nerek naturalnych.
Zmiany zachodzšce w ciężkiej sepsie w zakresie ilo?ci i jako?ci płynu pozakomórkowego oraz funkcji narzšdów, prowadzš do odmiennego rozdziału i dyspozycji leków. Antybiotyki hydrofilne i te wydalane przez nerki o mniejszej lipofilno?ci zagrożone sš ryzykiem występowania znacznych wahań dobowych w zakresie stężenia. Wynika to z faktu, że majš niskš Vd (<0,3-0,4 l kg-1; większo?ć aminoglikozydów, b-laktamów i glikopeptydów) i każda ucieczka płynu poza obszar naczyniowy obniża ich aktywno?ć. Logicznš konsekwencjš byłoby monitorowanie ich stężenia i dostosowanie indywidualnej dawki dla chorego. Przeciwnie, bardziej lipofilne antybiotyki, posiadajšce większš Vd (>1 l kg-1; większo?ć chinolonów i makrolidy), a których losy zależš głównie od funkcji wštroby i w mniejszym stopniu od sprawno?ci nerek, wykazujš większš stabilno?ć stężeń podczas terapii [6].
Rozważania dotyczšce farmakodynamiki
Dla antybiotyków parametry PD zwišzane sš z czynnikami PK w zakresie zdolno?ci tych leków do zabijania drobnoustrojów lub hamowania ich wzrostu. Na podstawie badań eksperymentalnych zidentyfikowano 3 parametry PD okre?lajšce profil działania różnych grup antybiotyków [14]:
1. Cmax/MIC – stosunek szczytowego stężenia leku uzyskanego po pojedynczej dawce – Cmax (mg l-1) do minimalnego stężenia hamujšcego MIC;
2. AUC24/MIC – stosunek pola pod krzywš zależno?ci zmian stężenia leku we krwi od czasu w cišgu 24 h – AUC24 (mg h-1 l-1) do MIC;
3. T>MIC (%) – czas, w którym stężenie leku we krwi pozostaje powyżej MIC.
Wska?nik AUC/MIC często służy do oceny skuteczno?ci działania antybiotyków zależnych od stężenia. Niektórzy autorzy używajš go jako wska?nika uniwersalnego [15].
Farmakodynamicznie, wielko?ć i natężenie aktywno?ci bakteriobójczej antybiotyków zależy od interakcji pomiędzy stężeniem leku w miejscu zakażenia, gęsto?ciš i fazš wzrostu drobnoustrojów oraz MIC [16]. Zmiana w zakresie któregokolwiek z tych czynników będzie oddziaływać na ich aktywno?ć przeciwdrobnoustrojowš i wynik leczenia. Z punktu widzenia farmakodynamicznego różne grupy antybiotyków majš różne typy działania przeciwdrobnoustrojowego w stosunku do drobnoustrojów (tab. III i ryc. 1).
Tab. III. Wła?ciwo?ci PD poszczególnych grup antybiotyków. Cmax – stężenie maksymalne antybiotyku we krwi; AUC24 – pole pod krzywš zależno?ci zmian stężenia leku we krwi od czasu w cišgu 24 h; T>MIC – czas, w którym stężenie antybiotyku w surowicy pozostaje powyżej MIC; Cmin – stężenie minimalne antybiotyku we krwi
| Antybiotyki | Profil PD | Optymalny parametr PD | Cel leczenia |
Aminoglikozydy
Fluorochinolony
Metronidazol | zależny od stężenia/przedłużony efekt | Cmax/MIC | zwiększyć stężenie
Cmax/MIC>10 |
Fluorochinolony
Azytromycyna
Tetracykliny
Glikopeptydy | zależny od stężenia wraz z zależno?ciš czasowš/przedłużony efekt | AUC24/MIC | zwiększyć stężenie i wydłużyć czas ekspozycji
AUC24/MIC>125 |
Penicyliny
Cefalosporyny
Aztreonam
Karbapenemy
Linezolid
Erytromycyna
Klarytromycyna
Klindamycyna
TMP/SMX | zależny od czasu/minimalnie przedłużony efekt | T>MIC | wydłużyć czas ekspozycji T>MIC=100%
Cmin/MIC 4-5 |
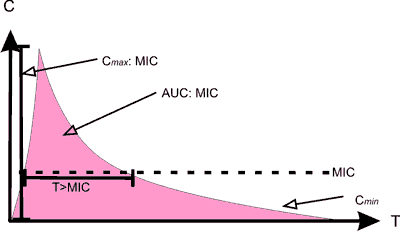
Ryc. 1. Parametry opisujšce PK/PD antybiotyków.
C – stężenie
T – czas
Cmax – stężenie szczytowe
AUC – pole pod krzywš
MIC – minimalne stężenie hamujšce
Cmin – stężenie minimalne
Dla zwiększenia skuteczno?ci terapii antybiotyki, których aktywno?ć jest zależna od stężenia (np. aminoglikozydy), należy podawać jeden raz na dobę w wysokich dawkach [17], aby uzyskać stosunek Cmax/MIC>8-10 [18, 19]. Dla antybiotyków o profilu działania zależnym od AUC (np. fluorochinolony), optymalnš warto?ć AUC/MIC osišga się stosujšc także wysokie dawki leków o długim t0,5 lub zwiększajšc często?ć dawkowania. Uważa się, że dla infekcji o mniejszym nasileniu oraz u chorych z prawidłowš odporno?ciš, warto?ć ta powinna wynosić 25-30 [20], a dla zakażeń wywołanych florš Gram-dodatniš: 40 [18, 21]. Dla ciężkich zakażeń oraz/lub u chorych z zaburzeniami w układzie immunologicznym adekwatne AUC/MIC oscyluje wokół 125 [20], natomiast w infekcjach o etiologii Gram-ujemnej optymalna terapia osišgana jest, gdy AUC/MIC, wg różnych autorów, waha się pomiędzy 125 a 500 [18, 22]. Większo?ć aminoglikozydów i fluorochinolonów wykazuje silny efekt poantybiotykowy (PAE – Postantibiotic Effect) zarówno wobec drobnoustrojów Gram-dodatnich jak i Gram-ujemnych [23], tak więc warto?ć stężenia minimalnego – Cmin (stężenie mierzone tuż przed podaniem kolejnej dawki) może znajdować się poniżej MIC [24].
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Pea F, Viale P, Furlanut M: Antimicrobial therapy in critically ill patients: a review of pathophysiological conditions responsible for altered disposition and pharmacokinetic variability. Clin Pharmacokinet 2005; 44: 1009-1034.
2. Hotchkiss RS, Karl IE:The pathophysiology and treatment of sepsis. N Engl J Med 2003; 348: 138-150.
3. Barger A, Fuhst C, Wiedemann B:Pharmacological indices in antibiotic therapy. J Antimicrob Chemother 2003; 52: 893-898.
4. Mehrotra R, De Gaudio R, Palazzo M: Antibiotic pharmacokinetic and pharmacodynamic considerations in critical illness. Intensive Care Med 2004; 30: 2145-2156.
5. Highet VS, Forrest A, Ballow C, Schentag JJ:Anibiotic dosing issues in lower respiratory tract infection: population-derived area under inhibitory curve is predictive of efficacy. J Antimicrob Chemother 1999; 43 (Suppl. A): 55-63.
6. Pinder M, Bellomo R, Lipman J:Pharmacological principles of antibiotic prescription in the critically ill. Anaesth Intensive Care 2002; 30: 134-144.
7. Wada DR, Drover DR, Lemmens HJ: Determination of the distribution volume that can be used to calculate the intravenous loading dose. Clin Pharmacokinet 1998; 35: 1-7.
8. De Paepe P, Belpaire FM, Buylaert WA: Pharmacokinetic and pharmacodynamic considerations when treating patients with sepsis and septic shock. Clin Pharmacokinet 2002; 41: 1135-1151.
9. Botha FJ, van der Bijl P, Seifart HI, Parkin DP: Fluctuation of the volume of distribution of amikacin and its effect on once-daily dosage and clearance in a seriously ill patient. Intensive Care Med 1996; 22: 443-446.
10. Buijk SL, Gyssens IC, Mouton JW, van Vliet A, Verbrugh HA, Bruining HA:Pharmacokinetics of ceftazidime in serum and peritoneal exudate during continuous versus intermittent administration to patients with severe intra-abdominal infections. J Antimicrob Chemother 2002; 49: 121-128.
11. Pea F, Porreca L, Baraldo M, Furlanut M: High vancomycin dosage regimens required by intensive care unit patients cotreated with drugs to improve haemodynamics following cardiac surgical procedures. J Antimicrob Chemother 2000; 45: 329-335.
12. Craig WA:Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial dosing of mice and men. Clin Infect Dis 1998; 26: 1-10.
13. Muller M, dela Pena A, Derendorf H: Issues in pharmacokinetics and pharmacodynamics of anti-infective agents: distribution in tissue. Antimicrob Agents Chemother 2004; 48: 1441-1453.
14. Roberts JA, Lipman J:Antibacterial dosing in intensive care: pharmacokinetics, degree of disease and pharmacodynamics of sepsis. Clin Pharmacokinet 2006; 45: 755-773.
15. Schentag JJ, Nix DE, Forrest A, Adelman MH: AUIC – the universal parameter within the constraint of a reasonable dosing interval. Ann Pharmacother 1996; 30: 1029-1031.
16. Nicolau DP:Optimizing outcomes with antimicrobial therapy through pharmacodynamic profiling. J Infect Chemother 2003; 9: 292-296.
17. Olsen KM, Rudis MI, Rebuck JA, Hara J, Gelmont D, Mehdian R, Nelson C, Rupp ME:Effect of once-daily dosing vs. multiple daily dosing of tobramycin on enzyme markers of nephrotoxicity. Crit Care Med 2004; 32: 1678-1682.
18. Scaglione F: Can PK/PD be used in everyday clinical practice. Int J Antimicrob Agents 2002; 19: 349-353.
19. Schentag JJ, Gilliland KK, Paladino JA: What have we learned from pharmacokinetic and pharmacodynamic theories? Clin Infect Dis 2001; 32 (Suppl. 1): S39-46.
20. Jacobs MR:Optimisation of antimicrobial therapy using pharmacokinetic and pharmacodynamc parameters. Clin Microbiol Infect. 2001; 7: 589-596.
21. Nightingale CH, Grant EM, Quintiliani R: Pharmacodynamics and pharmacokinetics of levofloxacin. Chemotherapy 2000; 46 (Suppl. 1): 6-14.
22. Wright DH, Brown GH, Peterson ML, Rotschafer JC: Application of fluoroquinolone pharmacodynamics. J Antimicrob Chemother 2000; 46: 669-683.
23. MacKenzie FM, Gould IM: The post-antibiotic effect. J Antimicrob Chemother 1993; 32: 519-537.
24. Odenholt I:Pharmacodynamic effects of subinhibitory antibiotic concentrations. Int J Antimicrob Agents 2001; 17: 1-8.
25. Mouton JW, Vinks AA:Is continuous infusion of beta-lactam antibiotics worthwhile? Efficacy and pharmacokinetics considerations. J Antimicrob Chemother 1996; 38: 5-15.
26. Craig W: Interrelationship between pharmacokinetics and pharmacodynamics in determining dosage regimens for broad-spectrum cephalosporins. Diagn Microbiol Infect Dis 1995; 22: 89-96.
27. Hanes S, Wood G, Herling V, Croce M, Fabian T, Pritchard E, Boucher B: Intermittent and continuous ceftazidime infusion for critically ill trauma patients. Am J Surg 2000; 179: 436-440.
28. Mohr J, Wanger A, Rex J: Pharmacokinetic/pharmacodynamic modeling can help guide targeted antimicrobial therapy for nosocomial gram-negative infections in critically ill patients. Diagn Microbiol Infect Dis 2004; 48: 125-130.
29. National Committee for Clinical Laboratory Standards. Performance standards for antimicrobial susceptibility testing, 12th informational supplement. Approved standard M100-S12. National Committee for Clinical Laboratory Standards, 2002; 22 (1).
30. MacGowan AP, Wise R: Establishing MIC breakpoints and the interpretation of in vitro susceptibility tests. J Antimicrob Chemother 2001; 48 (Suppl. S1): 17-28.
31. Estes L: Review of pharmacokinetics and pharmacodynamics of antimicrobial agents. Mayo Clin Proc 1998; 73: 1114-1122.
32. Warner S:Diagnostics+Therapy=Theranostics. Strategy requires teamwork, partnering, and tricky regulatory maneuvering. The Scientist 2004; 18: 38.
33. Amsden G, Duran M: Interpretation of antibacterial susceptibility reports: in vitro versus clinical break-points. Drugs 2001; 61: 163-166.
34. Ali MZ, Goetz MB:A meta-analysis of the relative efficacy and toxicity of single daily dosing versus multiple daily dosing of aminoglycosides. Clin Infect Dis 1997; 24: 796-809.
35. Munckhof WJ, Grayson ML, Turnidge JD: A meta-analysis of studies on the safety and efficacy of aminoglycosides given either once daily or as divided doses. J Antimicrob Chemother 1996; 37: 645-663.
36. Buijk SE, Mouton JW, Gyssens IC, Verbrugh HA, Bruining HA: Experience with a once-daily dosing program of aminoglycosides in critically ill patients. Intensive Care Med 2002; 28: 936-942.
37. Fantin B, Farinotti R, Thabaut A, Carbon C:Conditions for the emergence of resistance to cefpirome and ceftazidime in experimental endocarditis due to Pseudomonas aeruginosa. J Antimicrob Chemother 1994; 33: 563-569.
38. Pea F, Viale P:The antimicrobial therapy puzzle: Could pharmacokinetic-pharmacodynamic relationships be helpful in addressing the issue of appropriate pneumonia treatment incritically ill patiens? Clin Infect Dis 2006; 42: 1764-1771.
39. Turnidge JD:The pharmacodynamics of beta-lactams. Clin Infect Dis 1998; 27: 10-22.
40. Angus BJ, Smith MD, Suputtamongkol Y, Mattie H, Walsh AL, Wuthiekanun V, Chaowagul W, White NJ:Pharmacokinetic-pharmacodynamic evaluation of ceftazidime continuous infusion vs intermittent bolus injection in septicaemic melioidosis. Br J Clin Pharmacol 2000; 50: 184-191.
41. Craig W:The role of pharmacodynamics in effective treatment of community- acquired pathogens. Advanced Studies in Medicine 2002; 2: 126-134.
42. Lipman J, Wallis S, Rickard C:Low plasma cefepime levels in critically ill septic patients: pharmacokinetic modeling indicates improved troughs with revised dosing. Antimicrob Agents Chemother 1999; 43: 2559-2561.
43. Lipman J, Gomersall CD, Gin T, Joynt GM, Young RJ: Continuous infusion ceftazidime in intensive care: a randomized controlled trial. J Antimicrob Chemother 1999; 43: 309-311.
44. Kollef MH, Sherman G, Ward S, Fraser VJ: Inadequate antimicrobial treatment of infections: a risk factor for hospital mortality among critically ill patients. Chest 1999; 115: 462-474.
45. MacGowan A, Bowker K:Continous infusion of beta-lactam antibiotics. Clin Pharmacokinet 1998; 35: 391-402.
46. van Zanten AR, Polderman KH:Rational use of antibiotics in the ICU: Optimum efficacy for the lowest cost; in: Yearbook of Intensive Care and Emergency Medicine (Ed.: Vincent JL), Springer-Verlag, Berlin, Heidelberg and New York, 2005: 337-348.
47. Nicolau DP, McNabb J, Lacy MK, Quintiliani R, Nightingale CH:Continuous versus intermittent administration of ceftazidime in intensive care unit patients with nosocomial pneumonia. Int J Antimicrob Agents 2001; 17: 497-504.
48. Jaruratanasirikul S, Sriwiriyajan S, Punyo J:Comparison of the pharmacodynamics of meropenem in patients with ventilator-associated pneumonia following administration by 3-hour infusion or bolus injection. Antimicrob Agents Chemother 2005; 49: 1337-1339.
49. Lorente L, Lorenzo L, Martin MM, Jimenez A, Mora ML:Meropenem by continuous versus intermittent infusion in ventilator-associated pneumonia due to gram-negative bacilli. Ann Pharmacother 2006; 40: 219-223.
50. Zhou J, Dong Y, Zhao X, Lee S, Amin A, Ramaswamy S, Domagala J, Musser JM, Drlica K:Selection of antibiotic-resistant bacterial mutants: allelic diversity among fluoroquinolone-resistant mutations. J Infect Dis 2000; 182: 517-525.
51. Hyatt JM, Schentag JJ:Pharmacodynamic modeling of risk factors for ciprofloxacin resistance in Pseudomonas aeruginosa. Infect Control Hosp Epidemiol 2000; 21 (Suppl. 1): S9-11.
52. Lowdin E, Odenholt I, Cars O: In vitro studies of pharmacodynamic properties of vancomycin against Staphylococcus aureus and Staphylococcus epidermidis. Antimicrob Agents Chemother 1998; 42: 2739-2744.
53. Knudsen JD, Fuursted K, Raber S, Espersen F, Frimodt-Moller N: Pharmacodynamics of glycopeptides in the mouse peritonitis model of Streptococcus pneumoniae or Staphylococcus aureus infection. Antimicrob Agents Chemother 2000; 44: 1247-1254.
54. Rello J, Sole-Violan J, Sa-Borges M, Garnacho-Montero J, Munoz E, Sirgo G, Olona M, Diaz E:Pneumonia caused by oxacillin-resistant Staphylococcus aureus treated with glycopeptides. Crit Care Med 2005; 33: 1983-1987.
55. Wysocki M, Delatour F, Faurisson F, Rauss A, Pean Y, Misset B, Thomas F, Timsit JF, Similowski T, Mentec H, Mier L, Dreyfuss D:Continuous versus intermittent infusion of vancomycin in severe Staphylococcal infections: prospective multicenter randomized study. Antimicrob Agents Chemother. 2001; 45: 2460-2467.
56. Bodi M, Ardanuy C, Olona M, Castander D, Diaz E, Rello J:Therapy of ventilator-associated pneumonia: the Tarragona strategy. Clin Microbiol Infect 2001; 7: 32-33.