© Borgis - Postępy Nauk Medycznych 6/2010, s. 509-514
*Joanna Kołodziejczyk, Barbara Wachowicz
Rola układu fibrynolitycznego w progresji nowotworówa)
The fibrinolytic system in tumor progression
Katedra Biochemii Ogólnej, Uniwersytet Łódzki
Kierownik Katedry: prof. dr hab. Barbara Wachowicz
Streszczenie
Układ fibrynolityczny wraz z metaloproteinazami macierzy komórkowej tworzy złożony system enzymatyczny o szerokim zakresie proteolitycznego działania. Enzymy uczestniczące w fibrynolizie oraz metaloproteinazy są zaangażowane w przebieg wielu procesów fizjologicznych i patologicznych, których rozwój wymaga migracji komórek i przebudowy tkanek. Badania z ostatnich lat wskazują na aktywny udział układu fibrynolitycznego w progresji nowotworów, w której białka fibrynolizy odgrywają istotną rolę nie tylko dzięki bezpośredniej aktywności proteolitycznej, ale także poprzez regulację aktywności metaloproteinaz macierzy zewnątrzkomórkowej.
Summary
The fibrinolytic system cooperating with the extracellular matrix metalloproteinases constitutes a very complex enzymatic system, with a wide range of proteolytic activity. Fibrinolytic enzymes and metalloproteinases are involved in various physiological and pathological processes that require tissue remodeling and cell migration. A growing number of data confirming an important role of the fibrinolytic system in tumor progression and metastasis has been reported. Fibrinolytic system may participate directly in tumor progression by plasmin activity, or indirectly via metalloproteinases regulation as well.

Fibrynoliza stanowi w układzie hemostazy naturalną przeciwwagę dla procesu krzepnięcia krwi. Układ fibrynolityczny kontroluje ilość włóknika tworzonego z fibrynogenu w wyniku aktywacji krzepnięcia, usuwa niepotrzebną skrzeplinę i zapewnia utrzymanie płynności krwi. Wewnątrznaczyniowa proteoliza jest główną fizjologiczną funkcją układu fibrynolitycznego, ale białka uczestniczące w fibrynolizie są zaangażowane również w inne procesy biologiczne; wraz z metaloproteinazami macierzy zewnątrzkomórkowej (MMPs, ang. matrix metalloproteinases) tworzą złożony układ enzymatyczny odpowiedzialny za proteolizę zewnątrznaczyniową.
Zarówno fizjologiczne, jak i patologiczne procesy zachodzące w organizmie wiążą się z migracją komórek i zmianami w strukturze tkanek, co wymaga degradacji macierzy zewnątrzkomórkowej. Ze względu na szybkość procesu oraz inwazyjność, zmiany w tkankach i narządach są szczególnie istotne w progresji nowotworów. Inwazyjny charakter choroby stanowi jedną z głównych trudności w leczeniu nowotworów, gdyż metastazę umożliwia komórkom nowotworowym skomplikowany układ proteaz trawiący macierz zewnątrzkomórkową. W skład tego systemu wchodzą enzymy układu fibrynolitycznego, współdziałające z enzymami z grupy MMPs. Metaloproteinazy są czynnikiem umożliwiającym przebudowę tkanek i migrację komórek, a ich działanie jest powiązane i częściowo zależne od białek związanych z fibrynolizą. Wzrastająca liczba dostępnych danych wskazuje na różnorodne drogi zaangażowania białek fibrynolizy w rozwój nowotworów, m.in. poprzez działanie mitogenne i prozapalne. Ponadto, jako odpowiedzialny za aktywację metaloproteinaz układ fibrynolityczny uczestniczy w przebudowie tkanek i może odgrywać istotną rolę w progresji zmian nowotworowych.
Układ fibrynolityczny
Funkcjonowanie układu fibrynolitycznego opiera się na proteolitycznej aktywności plazminy, odpowiadającej przede wszystkim za usuwanie fibryny (włóknika), poprzez przekształcanie jej do produktów degradacji fibryny (FDP, ang. fibrin degradation products). Proenzym plazminy – plazminogen, ulega przekształceniu do plazminy na skutek działania aktywatorów (ryc. 1). Głównym fizjologicznym aktywatorem fibrynolizy jest t-PA, (aktywator plazminogenu typu tkankowego, ang. tissue-type plasminogen activator), aktywujący plazminogen w obecności powstałego włóknika. (1). Do aktywacji fibrynolizy dochodzi również w wyniku działania aktywatora plazminogenu typu urokinazowego (u-PA, ang. urokinase – type plasminogen activator). W warunkach fizjologicznych za aktywację fibrynolizy odpowiada głównie t-PA, natomiast u-PA jest w większym stopniu zaangażowany w proteolizę zwnątrznaczyniową (2).
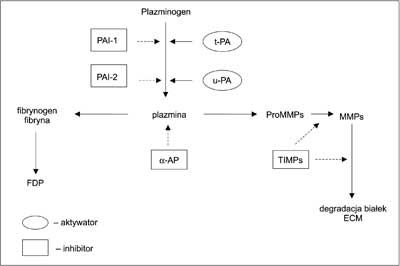
Ryc. 1. Schemat układu fibrynolitycznego (objaśnienia w tekście).
Działanie układu fibrynolitycznego jest kontrolowane poprzez inhibitory blokujące etap przekształcenia plazminogenu do plazminy, lub poprzez hamowanie już powstałej plazminy. Głównym inhibitorem aktywacji fibrynolizy jest inhibitor aktywatorów plazminogenu pochodzący z komórek śródbłonkowych naczyń krwionośnych (inhibitor typu 1, PAI-1; ang. plasminogen activator inhibitor – 1). PAI-1 jest syntetyzowany w komórkach śródbłonka ściany naczynia krwionośnego, komórkach wątroby i mięśni gładkich naczyń krwionośnych, skąd uwalniany jest do osocza i macierzy zewnątrzkomórkowej (3); część puli PAI-1 pochodzi z ziarnistości płytek krwi, z których jest uwalniany w procesie sekrecji w czasie tworzenia skrzepu (4). Czynnikami stymulującymi wydzielanie PAI-1 są cytokiny, m.in.: TGF-β, TNF-α, IL-1 i IL-6. Poziom PAI-1 ulega zmianom w czasie cyklu dobowego, jednak odwrotnie do rytmu wytwarzania t-PA. Poziom t-PA i PAI-1, a więc głównego aktywatora i głównego inhibitora fibrynolizy, odpowiada za aktywność fibrynolityczną krwi i przeciwzakrzepowy potencjał ściany naczyniowej, określany poprzez stosunek ilości PAI-1 do ilości t-PA związanego z komórkami śródbłonka (5). Inhibitor aktywatorów plazminogenu typu 2 (PAI-2, plasminogen activator inhibitor-2) pojawia się w osoczu kobiet w ciąży (6). Aktywność enzymatyczna plazminy regulowana jest przede wszystkim przez α2-antyplazminę (α2-AP) (7). Wybrane parametry charakteryzujące białka układu fibrynolitycznego przedstawiono w tabeli 1.
Tabela 1. Wybrane parametry charakteryzujące białka układu fibrynolitycznego.
| Białko | Główne miejsce syntezy | Stężenie w osoczu mg/ml | Czas pół-trwania |
| Plazminogen | wątroba | 200 | 2,2 dnia |
| Plazmina | - | - | - |
| t-PA | śródbłonek | 0,005 | " 5 min |
| u-PA | śródbłonek monocyty makrofagi | 0,008 | " 8 min |
| u-PAR | śródbłonek | - | - |
| a2-antyplazmina | wątroba | 70 | 2,6 dnia |
| PAI-1 | śródbłonek wątroba mięśnie gładkie ściany naczynia | 0,05 | 6 min |
| PAI-2 | łożysko | <0,005 | - |
Metaloproteinazy macierzy zewnątrzkomórkowej
Enzymy zaliczane do metaloproteinaz macierzy zewnątrzkomórkowej stanowią zróżnicowaną grupę cynko-zależnych endopeptydaz odpowiadających za hydrolizę składników macierzy zewnątrzkomórkowej (ang. extracellular matrix, ECM) (8). Zmiany w ECM i jej degradacja są niezbędne do przebudowy tkanek, zmieniających się zarówno w wyniku procesów fizjologicznych, jak i patologicznych (9). Ze względu na zróżnicowanie metaloproteinaz pod względem budowy i preferencji substratowych, wyodrębniono 5 grup tych enzymów: kolagenazy, żelatynazy, stromielizyny i metaloproteinazy błonowe (ang. membrane-type matrix metalloproteinases; MT-MMPs) i inne (matrylizyny) (10).
MMPs są syntetyzowane w formie zymogenów (proMMPs) głównie przez fibroblasty, komórki mięśni gładkich, komórki śródbłonka i miocyty (11), a za kontrolowanie ich aktywności proteolitycznej odpowiadają tkankowe inhibitory metaloproteinaz (ang. tissue inhibitors of matrix metalloproteinases, TIMPs) (12).
Współdziałanie układu fibrynolitycznego i metaloproteinaz
Plazmina może bezpośrednio degradować składniki macierzy, takie jak: proteoglikany, kolagen IV, laminina i fibronektyna. Zakres jej proteolitycznego działania na składniki ECM zwiększa się poprzez aktywację metaloproteinaz (13). Oddziaływanie białek fibrynolitycznych z metaloproteinazami może mieć charakter bezpośredni lub pośredni, ponieważ białka te uczestniczą w procesach aktywacji proMMPs. Plazmina w warunkach in vitro bezpośrednio aktywuje następujące prometaloproteinazy: proMMP-1, proMMP-3, proMMP-9, proMMP-10 i proMMP-13 (14), natomiast aktywacja proMMP-2 ma charakter pośredni i wymaga udziału innej metaloproteinazy (15).
Enzymy związane z fibrynolizą są także substratami dla metaloproteinaz. MMP-3, MMP-7, MMP-9 i MMP-12 mogą hydrolizować plazminogen, a MMP-12 rozszczepia jego cząsteczkę powodując powstanie fragmentu podobnego do angiostatyny (16). ProMMP-3 może wiązać t-PA oraz plazminogen i wpływać na efektywność aktywacji plazminogenu poprzez zwiększenie powinowactwa t-PA do plazminogenu (17).
Status fibrynolizy w chorobach nowotworowych
Zakrzepica jest najczęstszym powikłaniem choroby nowotworowej i drugą, po infekcjach, przyczyną zgonów chorych na nowotwory. Kliniczne objawy zakrzepicy pojawiają się u ok. 15% chorych na nowotwory złośliwe, a stwierdzenie zakrzepicy może być jednym ze wskaźników sugerujących rozwijanie się choroby nowotworowej (tzw. nowotwór utajony – niemy klinicznie) (18). W większości chorób nowotworowych obserwuje się tendencje prozakrzepowe, wynikające zarówno ze stymulacji układu krzepnięcia przez stan zapalny, czynniki aktywujące krzepnięcie krwi i aktywację płytek krwi, jak również będące wynikiem tłumienia aktywności układu fibrynolitycznego. Pobudzenie układu krzepnięcia krwi u pacjentów z nowotworami skutecznie osłabia fibrynolizę wewnątrznaczyniową, zwiększając prawdopodobieństwo wystąpienia powikłań zatorowo-zakrzepowych.
Tkanki zmieniono nowotworowo wytwarzają szereg czynników aktywujących krzepnięcie, m.in. IL-1β, czy TNF-α, a także stymulujących wytwarzanie przez komórki śródbłonka czynnika tkankowego (ang. tissue factor, TF), który jest głównym aktywatorem krzepnięcia (19). Same komórki nowotworowe również zdolne są do wytwarzania TF, a ponadto generują inny silny aktywator układu krzepnięcia, prokoagulant nowotworowy (ang. cancer procoagulant, CP) (20), powodując efektywne pobudzenie kaskady krzepnięcia krwi. Duża efektywność stymulacji krzepnięcia przez CP wynika z braku wrażliwości na hamujący wpływ fizjologicznych antykoagulantów obecnych w osoczu, takich jak: α1-antytrypsyna, α1-antychymotrypsyna, α2-makroglobulina, czy antytrombina III (21). Pobudzenie krzepnięcia może nasilać wydzielanie prokoagulantu, homologicznego do części antygenu HLA-DR ( human leukocyte antigen – jeden z ludzkich antygenów leukocytarnych odpowiadających MHC – major histocompatibility complex), a także PAA/PCA ( platelet aggregating activity/procoagulant activity) – aktywatora płytek krwi i czynnika X (22, 23). Zwiększenie ryzyka zakrzepicy jest także skutkiem ubocznym terapii przeciwnowotworowej. Chemioterapia indukuje wydzielanie koagulantów i cytokin z komórek nowotworowych, a jednocześnie działa cytotoksycznie na śródbłonek ściany naczynia osłabiając jego przeciwpłytkowe i przeciwzakrzepowe działanie (24).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Collen D, Lijnen HR: Tissue-type plasminogen activator: a historical perspective and personal account. J Thromb Haemost 2004; 2: 541-46.
2. Stepanova VV, Tkachuk VA: Urokinase as a multidomain protein and polyfunctional cell regulator. Biochemistry (Moscow) 2002; 67: 109-18.
3. Gils A, Declerck PJ: Plasminogen Activator Inhibitor-1. Curr Med Chem 2004; 11: 2323-34.
4. Czekay RP, Loskutoff DJ: Unexpected role of plasminogen activator inhibitor 1 in cell adhesion and detachment. Exp Biol Med 2004; 229: 1090-96.
5. Dobrovolsky AB, Titaeva EV: The fibrynolysis system: regulation of activity and physiologic functions of its amin components. Biochemistry (Moscow) 2002; 67: 116-26.
6. Yu H, Maurer F, Medcalf RL: Plasminogen activator inhibitor type 2: a regulator of monocyte proliferation and differentiation. Blood 2002; 99: 2810-18.
7. Turner RB, Liu L, Sazonova IY et al.: Structural Elements That Govern the Substrate Specificity of the Clot-dissolving Enzyme Plasmin. J Biol Chem 2002; 277: 33068-74.
8. Olszyński K, Zimowska M: Budowa i funkcje metaloproteinaz macierzy zewnątrzkomórkowej. Post Bioch 2009; 55 (1): 7684.
9. Johnson LL, Dyer R, Hupe DJ: Matrix metalloproteinases. Curr Opin Chem Biol 1998; 2: 466-71.
10. Ny T, Wahlberg P, Brändström IJ: Matrix remodeling in the ovary: regulation and functional role of the plasminogen activator and matrix metalloproteinase systems. Mol Cell Endocrinol 2002; 187: 29-38.
11. Hoit BD: Matrix metalloproteinases and atrial structural remodeling. J Am Coll Cardiol 2003; 42 (2): 345-47.
12. Brew K, Dinakarpandian D, Nagase H: Tissue inhibitors of metalloproteinases: evolution, structure and function. Biochim Biophys Acta 2000; 1477: 267-83.
13. Murphy G, Atkinon S, Ward R et al.: The role of plasminogen activators in the regulation of connective tissue metalloproteinases. Ann Acad Sci 1992; 667: 1-12.
14. Lijnen HR: Molecular interactions between the plasminogen/plasmin and matrix metalloproteinase systems. Fibrinolysis Proteol 2000; 14: 175-81.
15. Herouy Y: The role of matrix metalloproteinases (MMPs) and their inhibtors in venous leg ulcer healing. Phlebolymphology 2004; 44: 231-43.
16. Raza SL, Nehring LC, Shapiro SD et al.: Proteinase-activated receptor-1 regulation of macrophage elastase (MMP-12) secretion by serine proteinases. J Biol Chem 2000; 275: 41243-50.
17. Arza B, Holyaerts MF, Felez J et al.: Prostromelysin-1 (proMMP-3) stimulates plasminogen activation by tissue type plasminogen activator. Eur J Biochem 2000; 267: 6378-84.
18. Wojtukiewicz MZ, Rucińska M: Zakrzepica żył głębokich a nowotwory utajone. Deep venous thrombosis and occult cancer. Onkol Pol 1998; 2: 93-97
19. De Cicco M: The prothrombotic state in cancer: pathogenic mechanisms. Crit Rev Oncol Hematol 2004; 50: 187-96.
20. Nijziel MR, van Oerle R, Hillen HFP et al.: From Trousseau to angiogenesis: the link between the haemostatic system and cancer. Neth J of Med 2006; 64: 403-10.
21. Mielicki WP, Mielicka E, Gordon SG: Cancer procoagulant activity studies using synthetic chromogenic substrates. Thromb Res 1997; 87: 251-56.
22. Cavanaugh PG, Sloane BF, Bajkowski AS et al.: Purification and characterization of platelet aggregating activity from tumor cells: copurification with procoagulant activity. Thromb Res 1985; 37: 309-26.
23. Chelladurai M, Honn KV, Walz DA: HLA-DR is a procoagulant. Biochem Biophys Res Commun1991; 78: 467-73.
24. Karczmarek-Borowska B, Ładna E, Wójcik J et al.: Zator tętnicy płucnej w trakcie chemioterapii u chorego z nienasieniakowatym nowotworem jądra – opis przypadku. Pulmonary embolism during chemotherapy in patient with nonseminoma testicular cancer – case report. Onkol Pol 2003; 127-31.
25. Falanga A, Panova-Noeva M, Russo L: Procoagulant mechanisms in tumour cells. Best Pract Res Clin Haematol 2009; 22: 49-60.
26. Berger DH: Plasmin/plasminogen system in colorectal cancer. World J Surg 2002; 26: 767-71.
27. Binder BR, Mihaly J: The plasminogen activator inhibitor „paradox” in cancer. Immunol Lett 2008; 118: 116-14.
28. Ossowski L, Reich E: Antibodies to plasminogen activator inhibit human tumor metastasis. Cell 1983; 35: 611-19.
29. Durand MV, Bodker JS, Christensen A et al.: Plasminogen activator inhibitor-I and tumour growth, invasion, and metastasis. Thromb Haemost 2004; 91: 438-49.
30. Montuori N, Visconte V, Rossi G et al.: Soluble and cleaved forms of the urokinase-receptor: degradation products or active molecules? Thromb Haemost 2005; 93: 192-79.
31. Łojko A, Zawilska K, Grodecka-Gazdecka S, Komarnicki M: Zaburzenia hemostazy a nasilenie procesu neoangiogenezy u chorych na raka piersi. Relation between abnormalities of heamostasis and neoangiogenesis in breast cancer patients. Współcz Onkol 2006; 10: 515-20.
32. Shiba E, Kim SJ, Taguchi T et al.: A prospective study on the prognostic significance of urokinase-type plasminogen levels in breast cancer tissue. J Cancer Res Clin Oncol 1997; 123: 555-59.
33. Smith R, Xue A-Q, Gill A et al.: High expression of plasminogen activator inhibitor-2 (PAI-2) is a predictor of improved survival in patients with pancreatic adenocarcinoma. World Surg 2007; 31: 493-02.
34. Tan X, Egami H, Nozawa F et al.: Analysis of the invasion-metastasis mechanism in pancreatic cancer: Involvement of plasmin(ogen) cascade proteins in the invasion of pancreatic cancer cells. Int J Oncol 2006; 28: 369-74.
35. Diaz VM, Planagum J, Thompson TM et al.: Tissue plasminogen activator is required for the growth, invasion, and angiogenesis of pancreatic tumor cells. Gastroenterology 2002; 122: 806-19.
36. Baker EA, Leaper DJ: The plasminogen activator and matrix metalloproteinase systems in colorectal cancer: relationship to tumour pathology. Eur J Cancer 2003; 39: 981-88.
37. Schrohl AS, Christensen IJ, Pedersen AN et al.: Tumor tissue concentrations of the proteinase inhibitors tissue inhibitor of metalloproteinases-1 (TIMP-1) and plasminogen activator inhibitor type 1 (PAI-1) are complementary in determining prognosis in primary breast cancer. Mol Cell Proteomics 2003; 2: 164-72.
38. Yang S-F, Hsieh Y-S, Lin C-L et al.: Increased plasma levels of urokinase plasminogen activator and matrix metalloproteinase-9 in nonsmall cell lung cancer patients. Clin Chim Acta 2005; 354: 91-99.
39. Pryczynicz A, Guzińska-Ustymowicz K, Dymicka-Piekarska V et al.: Expression of matrix metalloproteinase 9 in pancreatic ductal carcinoma is associated with tumor metastasis formation. Folia Histochem Cytobiol 2007; 45: 37-40.
40. MacDougall J, Matrisian LM: Contributions of tumor and stromal matrix metalloproteinases to tumor progression, invasion and metastasis. Cancer Metastasis Rev 1995; 14: 351-62.
41. Ji F, Chen Y-L, Jin E-Y et al.: Relationship between matrix metalloproteinase-2 mRNA expression and clinicopathological and urokinase-type plasminogen activator system parameters and prognosis in human gastric cancer. World J Gastroenterol 2005; 11: 3222-26.
42. Khasigov PZ, Podobed OV, Gracheva TS et al.: Role of Matrix Metalloproteinases and their inhibitors in tumor invasion and metastasis. Biochemistry (Moscow) 2003; 68: 711-17.