© Borgis - Postępy Fitoterapii 3/2010, s. 131-146
*Tadeusz Wolski, Tomasz Baj, Agnieszka Ludwiczuk, Kazimierz Głowniak, Radosław Niedźwiecki
Analiza fitochemiczna korzeni hakorośli rozesłanej ( Harpagophytum procumbens DC.)
Phytochemical analysis of roots of devil's claw (Harpagophytum procumbens DC.)
Katedra i Zakład Farmakognozji z Pracownią Roślin Leczniczych, Uniwersytet Medyczny w Lublinie
Kierownik Katedry: prof. dr hab. Kazimierz Głowniak
Summary
In the present paper the phytochemical analysis of Devil's Claw (Harpagophytum procumbens DC.) roots concerning analysis of iridoid derivatives, flavonoids, phenolic acids and tannins was described. The content of the major active compound, harpagoside, in the plant material and pharmaceutical preparation named Pagosid, was determined. Quantitative analysis was performed by use of spectrophotometric, TLC and HPLC methods. The content of harpagoside in Harpagophyti radix amounted 1.25% (determined by spectrophotometric method), 1.29% (TLC) and 2.17% (HPLC), while in tablets Pagosid 0.77%, 0.99% and 0.87%, respectively. These data indicated that Devil's Claw roots meet the requirements of European Pharmacopoeia (not less than 1.2% of harpagoside) and DAB 10 (not less than 1%). The content of harpagoside in Pagosid was below both pharmacopoeias requirements. Qualitative analysis of the flavonoids did not show the presence of free aglycones in plant material, while the presence of luteolin, kaempherol and flavone glycosides was confirmed. The total content of flavonoids in Harpagophyti radix recalculated into quercetin was 0.01%. Analysis of phenolic acids showed the presence of cinnamic, ferulic, p-coumaric, chlorogenic, caffeic and protocatechuic acids. Additionally, gallic acid was identified in the fraction received after hydrolysis of Devil's Claw roots. The content of tannins in the investigated raw material amounted 5.53% recalculated into dry matter. Qualitative analysis of this group of compounds showed the absence of catechin derivatives, while the presence of gallotannins and caffeic acid derivatives was confirmed. It is worthy of mention that spectrophotometric determination of the harpagoside content using Godin reagent at 540 nm have been applied for the first time. Described method can be used for screening determination of the content of harpagoside in plant material and pharmaceutical preparations.

W poprzedniej pracy (1) przedstawiono przegląd piśmiennictwa dotyczącego hakorośli rozesłanej Harpagophytum procumbens DC. oraz opisano wygląd anatomiczno-morfologiczny surowca, a także omówiono skład fitochemiczny. Do głównych związków biologicznie aktywnych występujących w badanym surowcu należą irydoidy właściwe typu aukubiny, tj. harpagozyd (0,1-3%) oraz produkty jego rozpadu: 8-p-kumaroiloharpagid i harpagid. Ponadto występują: prokumbid i jego 6' p -kumarylowy ester, a także prokumbozyd. Ta grupa związków decyduje o wielokierunkowym działaniu farmakologicznym hakorośli. Surowiec ten zyskuje na znaczeniu i zastosowaniu w fitoterapii, czego dowodem jest prezentowana monografia w ostatniej edycji ESCOP (2).
Celem niniejszej pracy było wykonanie badań nad składem chemicznym korzeni hakorośli rozesłanej z gatunku Harpagophytum procumbens DC., należącej do rodziny Połapkowatych (Pedaliaceae). Badania te obejmowały analizę wstępną surowca, a więc oznaczenie suchej masy, składników mineralnych (popiołu), a także izolację, identyfikację i oznaczenie ilościowe głównych grup związków biologicznie aktywnych obecnych w surowcu: pochodnych irydoidów, flawonoidów, fenolokwasów i garbników. Ponadto przeprowadzono analizę zawartości głównego składnika czynnego – harpagozydu w badanym surowcu oraz preparacie Pagosid zawierającym wyciąg z korzenia Harpagophytum.
Materiał i metody
Materiał do badań stanowiły drugorzędowe korzenie (bulwy) hakorośli rozesłanej – Harpagophyti radix oraz preparat handlowy Pagosid firmy Dr Dünner (Szwajcaria). Badane surowce pochodziły z 2001 roku.
Metodyka badań
Analiza wstępna badanego surowca
Analiza wstępna obejmowała oznaczenie straty masy po suszeniu oraz zawartości substancji mineralnych (popiołu) w korzeniu Harpagophytum procumbens DC. Analizy te prowadzono zgodnie z FP V (3), a uzyskane wyniki przedstawiono w tabeli 1.
Tabela 1. Procentowa zawartość wilgoci i popiołu w korzeniu hakorośli rozesłanej ( Harpagophytum procumbens DC.).
| Nr próby | Wilgotność (% wag.) | Popiół (% wag.) |
1
2
3 | 10,48
10,42
10,49 | 5,56
5,50
5,32 |
| Średnia | 10,46 | 5,46 |
Analiza fitochemiczna badanego surowca i preparatu Pagosid
Przygotowanie ekstraktów do badań
Odważono 2,0 g rozdrobnionego surowca i przeniesiono do kolby okrągłodennej. Surowiec traktowano 20 ml metanolu i ogrzewano przez 30 min na łaźni wodnej pod chłodnicą zwrotną. Wyciąg przesączono przez watę do kolby miarowej o poj. 50 ml. Ekstrakcję wykonano ponownie, wyciągi połączono i uzupełniono metanolem do kreski. Wyciągi poddano dalszym analizom.
Ekstrakcję tabletek Pagosid przeprowadzono tą samą metodą, a do oznaczenia użyto jednej tabletki (1 tabl. odpowiada 820 mg korzenia Harpagophytum).
Wstępna analiza ekstraktu metodą chromatografii cienkowarstwowej (TLC)
Analizę TLC prowadzono na płytkach pokrytych żelem krzemionkowym Si-60 G F254. Chromatogramy rozwijano w układzie: 1-propanol:toluen:kwas octowy:woda (5:4:2:2 v/v).
Po rozwinięciu i wysuszeniu chromatogramy umieszczano pod lampa UV (λ=254 nm). Następnie płytki wywołano przez spryskanie odczynnikami:
– odczynnikiem Godina (A: 1% etanolowy roztwór waniliny, B: 5% etanolowy roztwór H2SO4),
– odczynnikiem EP (0,25 g aldehydu 4,4-dimetyloaminobenzoesowego rozpuszczono w mieszaninie 45 ml kwasu octowego lodowatego, 5 ml kwasu fosforowego 85% i 45 ml wody), całość ogrzewano 3-5 minut w temperaturze 100°C.
Wyniki przedstawiono w tabelach 2 i 3.
Tabela 2. Wartosci RF i barwy plam widocznych na chromatogramie pod lampą UV (λ=254 nm) i po spryskaniu odczynnikiem EP.
| Przedmiot badania | Plama | Barwa plamy w świetle UV (l=254 nm) | Barwa plamy po spryskaniu odczynnikiem EP | RF |
| Wzorzec harpagozydu | 1 | brunatna | jasnoniebieska | 0,61 |
| Ekstrakt z surowca | IA
IB
IC
ID
IE | ciemnożółta
ciemnożółta
ciemnożółta
ciemnożółta
brunatnoniebieska | biała
biała
biała
biała
jasnoniebieska | 0,20
0,24
0,41
0,50
0,62 |
| Ekstrakt z tabletek Pagosid | IIA
IIB
IIC
IID
IIE | ciemnożółta
ciemnożółta
ciemnożółta
ciemnożółta
brunatnoniebieska | biała
biała
biała
biała
jasnoniebieska | 0,20
0,26
0,42
0,50
0,61 |
Tabela 3. Wartosci RF i barwy plam na chromatogramie po spryskaniu odczynnikiem Godina.
| Przedmiot badania | Plama | Barwa plamy po spryskaniu odczynnikiem Godina | RF |
| Wzorzec harpagozydu | 1 | jasnoczerwona | 0,61 |
| Ekstrakt z surowca | IA
IB
IC
ID
IE | brunatna
brunatna
brunatna
brunatna
jasnoczerwona | 0,20
0,24
0,39
0,48
0,62 |
| Ekstrakt z tabletek Pagosid | IIA
IIB
IIC
IID
IIE | brunatna
brunatna
brunatna
brunatna
jasnoczerwona | 0,20
0,25
0,40
0,49
0,61 |
Oznaczenia jakościowe i ilościowe harpagozydu
Metoda spektrofotometryczna
Wyznaczanie analitycznej długości fali
Na płytkę pokrytą żelem krzemionkowym Si-60 G F254 naniesiono 0,01 ml, 0,02 ml i 0,04 ml ekstraktu z surowca i 0,02 ml wzorca harpagozydu o stężeniu 0,02%. Płytkę rozwinieto w układzie: 1-propanol:toluen:kwas octowy:woda (5:4:2:2 v/v).
Pod lampą UV (λ=254 nm) obrysowano plamy harpagozydu i przeniesiono preparatywnie do próbówek wirówkowych, dodano 4 ml odczynnika Godina. Próbówki ogrzewano na łaźni wodnej przez 5 min. Następnie wirowano przez 10 min, po czym zmierzono absorbancję próbek w zakresie długości fali 400-630 nm, stosując jako odnośnik odczynnik Godina, wyznaczając w ten sposób maksimum absorbancji, która wynosiła 540 nm (ryc. 1).
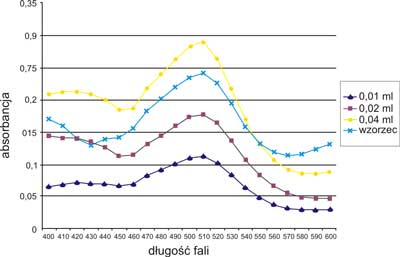
Ryc. 1. Zależność absorbancji od długości fali dla barwnego połączenia harpagozydu z odczynnikiem Godina.
Wyznaczanie krzywej wzorcowej harpagozydu
Na płytkę TLC naniesiono 25, 50, 75, 100 i 125 μl roztworu wzorca harpagozydu (0,02%) odpowiadające 5, 10, 15, 20 i 25 μg harpagozydu. Płytkę rozwinięto, a plamy harpagozydu przeniesiono ilościowo do próbówek wirówkowych. Do każdej z probówek dodano po 4 ml odczynnika Godina i ogrzewano na łaźni wodnej przez 10 min. Następnie odwirowano żel krzemionkowy i zmierzono absorbancję przy długości fali λ=540 nm. Wyniki przedstawiono na rycinie 2.
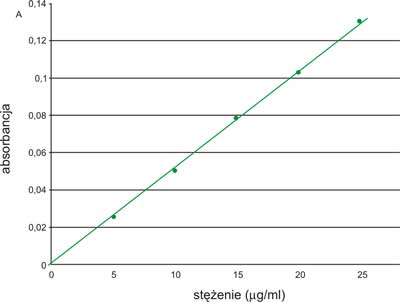
Ryc. 2. Zależność absorbancji od stężenia dla barwnego połączenia harpagozydu z odczynnikiem Godina (λ=540 nm).
Oznaczanie zawartości
Badane ekstrakty nanoszono na płytki TLC w ilości po 50 μl w trzech powtórzeniach. Dalej postępowano jak przy wyznaczaniu analitycznej długości fali. Zawartość harpagozydu wyznaczono na podstawie krzywej wzorcowej. Wyniki przedstawiono w tabeli 4.
Tabela 4. Wartość absorbancji barwnego połączenia harpagozydu z odczynnikiem Godina w badanym surowcu i w tabletkach Pagosid.
| Przedmiot badania | Numer próbki | Absorbancja | Średnia |
| Surowiec | 1
2
3 | 0,118
0,112
0,114 | 0,115 |
| Tabletki | 1
2
3 | 0,066
0,062
0,065 | 0,064 |
Metoda TLC
Przy pomocy automatycznego urządzenia do nanoszenia próbek Autosampler TLC III (Camag, Szwajcaria) naniesiono na płytkę pokrytą żelem krzemionkowym Si-60 G F254 po 10 μl ekstraktów z surowca i tabletek oraz 10, 20, 30, 40 i 50 μl wzorca o stężeniu 0,02%, co odpowiada 2, 4, 6, 8 i 10 μg harpagozydu. Płytkę rozwinęto na dystansie 15 cm w układzie: 1-propanol:toluen:kwas octowy:woda (5:4:2:2 v/v).
Następnie wykonano zdjęcie płytki w świetle UV (λ=254 nm) używając do tego celu VideoScanera TLC/HPTLC firmy Camag (Szwajcaria). Wykonane zdjęcie poddano obróbce przy pomocy programu VideoStore, otrzymując chromatogramy TLC dla ekstraktów z surowca i tabletek oraz wzorca o wyżej podanych stężeniach. Zawartość harpagozydu wyliczono na podstawie krzywej wzorcowej f=S(c), gdzie S – pole powierzchni piku wzorca; c – steżęnie wzorca (ryc. 3 i 4).

Ryc. 3. Chromatogram TLC wzorca harpagozydu o różnych stężeniach oraz wyciągu z surowca i tabletek Pagosid otrzymany w warunkach: Si-60 G F254/1-propanol:toluen:kwas octowy:woda (5:4:2:2 v/v); λ=254 nm.
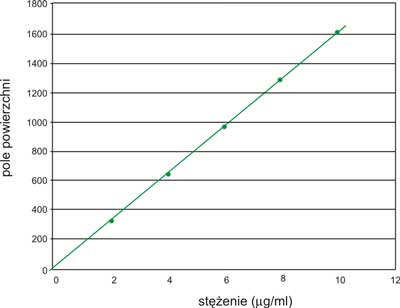
Ryc. 4. Zależność pola powierzchni piku od stężenia harpagozydu F=S(c).
Metoda HPLC
Analizę przeprowadzono zgodnie z metodą opisaną w Farmakopei Europejskiej (3. edycja) (4).
Przygotowanie wzorca: 2 mg harpagozydu rozpuszczono w 5 ml metanolu. Roztwór przeniesiono ilościowo do kolby miarowej o pojemności 10 ml i uzupełniono do kreski metanolem.
Standard wewnętrzny: 0,130 g cynamonianu metylu rozpuszczono w 50 ml metanolu i uzupełniono do 100 ml metanolem.
Przygotowanie próbki: Do 0,5 g sproszkowanego surowca dodano 50 ml metanolu. Wytrząsano 1 godz., następnie prząsaczono. Sączek wraz z pozostałością przeniesiono do kolby o poj. 100 ml, dodano 50 ml metanolu i ogrzewano pod chłodnicą zwrotną przez 1 godz., następnie ochłodzono i przesączono. Kolbę i sączek przemyto dwukrotnie po 5 ml metanolu. Filtraty i roztwory po przemyciu kolby i sączka połączono i odparowano do sucha pod zmniejszonym ciśnieniem w temperaturze nieprzekraczającej 40°C. Pozostałość ekstrahowano trzykrotnie porcjami po 5 ml metanolu i przesączono do kolbki na 25 ml. Po przemyciu sączka uzupełniono kolbę do kreski metanolem. Do 10 ml tego roztworu dodano 1 ml standardu wewnętrznego i rozcieńczono otrzymany roztwór do 25 ml metanolem. Identyczną procedurę zastosowano w przypadku preparatu Pagosid.
Analizę HPLC prowadzono stosując chromatograf typu LaChrom-Merck z detektorem diodowym DAD L-7450, pompą HPLC L-7100, dozownikiem Rheodyne, pętlą dozującą 10 μl, kolumną LiChrospher 100 RP-C18 o wymiarach 250 mm x 4 mm i średnicy ziaren 5 μm. Fazą ruchomą był metanol:woda (60:40 v/v), szybkość przepływu wynosiła 0,8 ml/min. Zawartość harpagozydu wyliczono według wzoru:
X = (m2 x F1 x 7,622)/(F2 x m1);
gdzie:
m1 – masa odważki (g),
m2 – masa cynamonianu metylu zawartego w 100 ml standardu wewnętrznego (g),
F1 – powierzchnia pod pikiem harpagozydu,
F2 – powierzchnia pod pikiem cynamonianu metylu.
Chromatogramy HPLC, będące efektem oznaczenia ilościowego harpagozydu, przedstawione są na rycinach 5-7, zaś porównawcze wyniki oznaczenia tego związku trzema metodami podane są w tabeli 5 oraz rycinie 8.
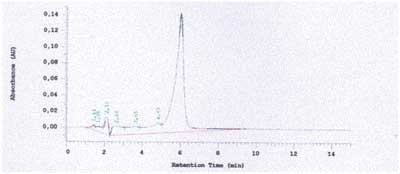
Ryc. 5. Chromatogram HPLC wzorca harpagozydu.
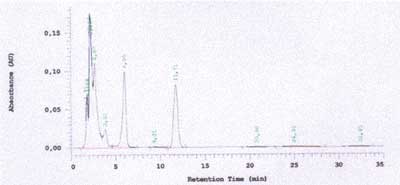
Ryc. 6. Chromatogram HPLC wyciągu z Harpagophyti radix z dodatkiem wzorca wewnętrznego.

Ryc. 7. Chromatogram HPLC wyciągu z tabletek Pagosid z dodatkiem wzorca wewnętrznego.
Tabela 5. Zawartość procentowa harpagozydu w wyciągach z korzenia i tabletek Pagosid oznaczona trzema metodami: spektrofotometryczną, TLC i HPLC.
| Przedmiot badań | Zawartość (%) | Zawartość (%) w przeliczeniu na suchą masę |
| A | B | C | A | B | C |
| Surowiec | 1,132 | 1,165 | 1,964 | 1,250 | 1,287 | 2,169 |
| Tabletki | 0,765 | 0,991 | 0,866 | 0,845 | 1,095 | 0,975 |
| A - Metoda spektrofotometryczna; B - Metoda densytometryczna; C - Metoda HPLC |
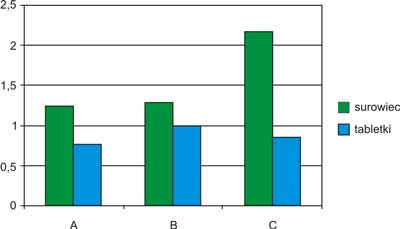
Ryc. 8. Wykres zależności zawartości harpagozydu w surowcu i tabletkach Pagosid od metody oznaczenia.
A – metoda spektrofotometryczna; B – metoda densytometryczna; C – metoda HPLC
Analiza flawonoidów
Oznaczanie ogólnej zawartości flawonoidów w surowcu
Oznaczenie przeprowadzono zgodnie z metodyką opisaną w FP V (3). Do analizy odważano dokładnie 1,0 g badanego surowca. Wyniki przedstawiono w tabeli 6.
Tabela 6. Zawartość procentowa flawonoidów w Harpagophyti radix w przeliczeniu na kwercetynę.
| Numer próby | Absorbancja | Zawartość flawonoidów (%) | Zawartość flawonoidów w przeliczeniu na suchą masę |
1
2
3 | 0,010
0,009
0,011 | 0,00875
0,007875
0,009625 | 0,0098
0,0088
0,0107 |
| Średnia | 0,0095 | 0,00875 | 0,0098 |
Analiza TLC flawonoidów
50,0 g rozdrobnionego korzenia hakorośli rozesłanej ekstrahowano eterem naftowym w aparacie Soxhleta w czasie około 10 godz. Surowiec pozbawiony ciał balastowych wysuszono, a następnie ekstrahowano metanolem. Ekstrakt metanolowy zagęszczono na wyparce obrotowej. Następnie wytrawiono niewielką ilością gorącej wody, pozostawiono na 24 godz. w lodówce, po czym przesączono. Ekstrakt wodny ekstrahowano octanem etylu, osuszono bezwodnym siarczanem sodu i przeprowadzono analizę TLC wobec wzorców na płytkach pokrytych poliamidem w układzie metanol:woda:kwas octowy (20:12,5:1 v/v) oraz na płytkach pokrytych żelem krzemionkowym Si-60 G F254 w układzie:toluen:mrówczan etylu:kwas mrówkowy (5:4:1 v/v).
Po rozwinięciu i wysuszeniu chromatogramy umieszczono pod lampą UV (λ=365 i 254 nm). Po czym spryskano płytki 5% metanolowym roztworem chlorku glinu.
Badaniom TLC poddano również ekstrakt otrzymany według procedury zastosowanej do oznaczenia ogólnej zawartości flawonoidów w surowcu. Ekstrakt ten odparowano do sucha, rozpuszczono w niewielkiej ilości metanolu i przesączono. Otrzymany roztwór nanoszono na płytki pokryte żelem krzemionkowym Si-60 G F254 wobec wzorców i rozwijano w układzie toluen:mrówczan etylu:kwas mrówkowy (5:4:1 v/v).
Po rozwinięciu i wysuszeniu chromatogramy umieszczono pod lampą UV (λ=254 nm) i zaznaczono plamy flawonoidów. Następnie, w celu oczyszczenia frakcji, żel wraz z plamami przenoszono do próbówek i ekstrahowano metanolem. Roztwory frakcjonowanych flawonoidów wraz z wzorcami nanoszono na płytkę i przeprowadzono analizę TLC analogiczną do wyżej opisanej.
Po rozwinięciu i wysuszeniu chromatogram umieszczono pod lampą UV (λ=254 nm) i wykonano zdjęcie za pomocą VideoScanu firmy CAMAG. Wartości Rf flawonoidów przedstawiono w tabeli 7.
Tabela 7. Wartości współczynników Rf dla poszczególnych plam flawonoidów obecnych na chromatogramie TLC.
| Przedmiot badania | Plama | Rf |
Flawon wzorzec
Surowiec
Luteolina wzorzec
Surowiec
Kemferol wzorzec
Surowiec
| 1
Ia
2
IIa
IIb
IIc
3
IIIa
IIIb | 0,68
0,67
0,45
0,39
0,45
0,47
0,55
0,55
0,57 |
Analiza fenolokwasów
Izolacja zespołu wolnych fenolokwasów
50,0 g korzenia hakorośli rozesłanej ekstrahowano eterem naftowym w aparacie Soxhleta przez około 10 godz. Następnie gilzy osuszono i przeprowadzono dalszą ekstrakcję metanolem. Ekstrakt metanolowy zagęszczono przez odparowanie rozpuszczalnika na wyparce obrotowej, a pozostałość wytrawiono gorącą wodą i pozostawiono w lodówce na dobę. Otrzymany ekstrakt wodny przesączono i ekstrahowano 10-krotnie eterem etylowym w porcjach po 50 ml. Ekstrakt eterowy zagęszczono i wytrząsano z 20 ml 5% roztworu wodorowęglanu sodowego. Warstwę wodno-węglanową zakwaszono kwasem solnym do pH=3 i ponownie ekstrahowano eterem etylowym (10 x 20 ml). Ekstrakt eterowy osuszono bezwodnym siarczanem sodowym i przesączono. Rozpuszczalnik odparowano, a suchą pozostałość rozpuszczono w 10 ml metanolu.
Izolacja zespołu fenolokwasów po hydrolizie surowca
Odważono 2,0 g rozdrobnionego surowca i umieszczono w kolbie okrągłodennej. Próbkę zalano 50 ml 4% roztworu kwasu siarkowego i umieszczono na łaźni wodnej pod chłodnicą zwrotną. Po 3 godz. wyciąg przesączono i ekstrahowano 10-krotnie eterem etylowym w porcjach po 10 ml. Wyciąg eterowy osuszono bezwodnym siarczanem sodowym i przesączono. Rozpuszczalnik odparowano, a suchą pozostałość rozpuszczono w 10 ml metanolu.
Analiza TLC
Analizie TLC poddano zarówno ekstrakt zawierający wolne fenolokwasy, jak i roztwór fenolokwasów po hydrolizie.
Wyciągi oraz wzorce naniesiono na płytkę TLC pokrytą żelem krzemionkowym Si-60 G F254 i rowijano w układzie: toluen:mrówczan etylu:kwas mrówkowy (5:4:1 v/v).
Po rozwinięciu i wysuszeniu chromatogram umieszczono pod lampą UV (λ=254 nm) i wykonano zdjęcie za pomocą VideoScanu Firmy CAMAG. Następnie płytki spryskano 1% metanolowym roztworem chlorku żelazowego i wykonano zdjęcie w swietle widzialnym. Wyniki przedstawiono na rycinach 9 i 10 oraz w tabeli 8.

Ryc. 9. Chromatogram TLC fenolokwasów obecnych w Harpagophyti radix po wywołaniu w świetle UV (λ=254 nm).
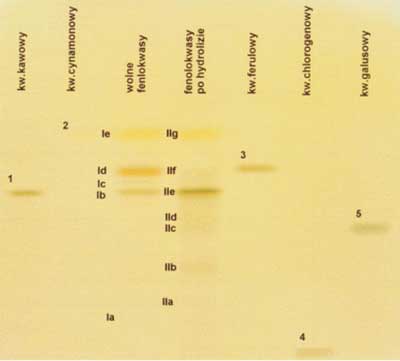
Ryc. 10. Chromatogram TLC fenolokwasów obecnych w Harpagophyti radix po wywołaniu 1% metanolowym roztworem FeCl3.
Tabela 8. Wartości współczynników RF dla poszczególnych plam fenolokwasów obecnych na chromatogramach przedstawionych na rycinach 9 i 10.
| Roztwór naniesiony | Plama | RF |
Kwas kawowy
Kwas cynamonowy
Ekstrakt z surowca zawierający frakcję wolnych fenolokwasów
Ekstrakt z surowca zawierający frakcję fenolokwasów po hydrolizie 4% H2SO4
Kwas ferulowy
Kwas chlorogenowy
Kwas galusowy | 1
2
Ia
Ib
Ic
Id
Ie
IIa
IIb
IIc
IId
IIe
IIf
IIg
3
4
5 | 0,51
0,68
0,09
0,51
0,53
0,56
0,68
0,13
0,31
0,40
0,43
0,51
0,56
0,68
0,56
0,04
0,40 |
Wykonano również analizę TLC fenolokwasów na płytkach pokrytych celulozą w układzie rozwijającym: woda:dioksan (10:1 v/v). Po rozwinięciu i wysuszeniu płytki wywołano przez spryskanie 1% metanolowym roztworem FeCl3. Wyniki podaje tabela 9.
Tabela 9. Wyniki analizy TLC fenolokwasów na płytkach pokrytych celulozą w układzie rozwijającym: woda:dioksan (10:1 v/v) po wywołaniu 1% metanolowym roztworem FeCl3.
| Przedmiot badania | Plama | Barwa plamy po spryskaniu 1% FeCl3 | RF |
Kwas kawowy
Kwas p-kumarowy
Kwas chlorogenowy
Fenolokwasy wolne
Fenolokwasy po hydrolizie
|
1
3
4
Ia
Ib
Ic
Id
IIa
IIb
IIc
IId
IIe
IIf
IIg
| ciemnobrunatna
pomarańczowo-brunatna
zielono-brunatna
ciemnobrunatna
ciemnobrunatna
ciemnobrunatna
pomarańczowo-brunatna
ciemnobrunatna
jasnobrunatna
niebiesko-brunatna
ciemnobrunatna
ciemnobrunatna
zielono-brunatna
pomarańczowo-brunatna |
0,24
0,83
0,77
0,49
0,69
0,77
0,85
0,23
0,33
0,45
0,61
0,69
0,76
0,86
|
Metoda HPLC
Ekstrakty otrzymane zgodnie z procedurą opisaną w procesie izolacji zespołu fenolokwasów poddano analizie HPLC.
Analizę przeprowadzono stosując chromatograf typu LaChrom-Merck z detektorem diodowym DAD L-7450, pompą HPLC L-7100, pętlą dozującą 20 μl, dozownikiem Rheodyne, kolumną LiChrospher 100 RP-C18 o wymiarach 250 mm x 4 mm i średnicy ziaren 5 μm. Faza ruchoma metanol + woda (25+75 v/v) z dodatkiem 1% v/v kwasu octowego; szybkość przepływu wynosiła 0,8 ml/min, a wielkość nastrzyku 20 μl. Identyfikację fenolokwasów przeprowadzono porównując ich czasy retencji (Rt) z wzorcami oraz spektroskopowo wyznacząjac ich widma DAD w zakresie długości fali 219-400 nm.
Wyniki przedstawiono na rycinach 11-12 oraz w tabelach 10 i 11.

Ryc. 11. Chromatogram HPLC wolnych fenolokwasów obecnych w Harpagophyti radix.
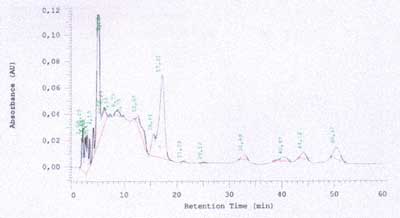
Ryc. 12. Chromatogram HPLC wolnych fenolokwasów otrzymanych po hydrolizie surowca.
Tabela 10. Wartości czasów retencji Rt dla wzorców kwasów fenolowych otrzymane w wyniku analizy HPLC.
| Fenolokwas | Czas retencji Rt (min) |
Kwas galusowy
Kwas protokatechowy
Kwas chlorogenowy
Kwas p-hydroksybenzoesowy
Kwas syryngowy
Kwas wanilinowy
Kwas kawowy
Kwas p-kumarowy
Kwas ferulowy |
4,46
6,81
9,96
10,43
11,93
13,86
15,37
28,77
35,39
|
Tabela 11. Wyniki analizy HPLC wolnych fenolokwasów i fenolokwasów obecnych w ekstrakcie po hydrolizie surowca.
| Przedmiot badania | Rt | Związek | Wsp. korelacji z widmem wzorca |
| Wyciąg z surowca zawierający wolne fenolokwasy | 6,41
15,03
27,50
34,40 | kw. protokatechowy
kw. kawowy
kw. p-kumarowy
kw. ferulowy | 0,9947
0,9955
1,0000
0,9997 |
| Wyciąg z surowca zawierający fenolokwasy po hydrolizie 4% H2SO4 | 17,14
32,53
40,35 | kw. kawowy
kw. p-kumarowy
kw. ferulowy | 0,9998
0,9940
0,9915 |
Analiza garbników
Sporządzanie ekstraktu z surowca
Oznaczenie wykonano metodą miareczkowo-wagową opisaną w FP IV (5). Odważono 10,0 g korzenia hakorośli rozesłanej, zalano 500 ml wody i powoli ogrzano do wrzenia. Ostudzony ekstrakt wodny przesączono przez zwitek waty do kolby miarowej o pojemności 1000 ml, starając się, aby surowiec nie spłynął na watę. Surowiec zalano ponownie 300 ml wody, ogrzano do wrzenia i gotowano przez 10 min. Po ochłodzeniu ekstrakt przesączono przez watę do kolby miarowej i uzupełniono wodą do kreski. Otrzymany ekstrakt przesączono przez bibułę, odrzucając pierwsze 20 ml przesączu.
Oznaczanie ilościowe garbników w Harpagophyti radix
Do zlewki o pojemności 150 ml odmierzono dokładnie 50 ml ekstraktu oraz 25 ml 0,1 M roztworu octanu miedziowego i zamieszano dokładnie bagietką. Wytrącony osad garbnikanów miedzi po upływie 12 godz. odsączono na uprzednio wysuszonym i zważonym sączku o średnicy 11 cm. Przezroczysty przesącz odstawiono. Resztki osadu spłukano na ten sam sączek i osad przemyto wodą, aż do ujemnej reakcji na jony miedzi (1 ml przesączu po dodaniu 1 ml 5% roztworu żelazocyjanku potasowego nie powinien wykazywać brunatnego zabarwienia). Sączek z osadem wysuszono w temperaturze 100-102°C do stałego ciężaru. Z odstawionego przesączu, zawierającego niezwiązany octan miedziowy, odmierzono dokładnie 25 ml do kolby stożkowej o pojemności 200 ml, dodano 10 ml 10% kwasu siarkowego, 2 g jodku potasu rozpuszczonego w 5 ml wody i 2 ml roztworu skrobi. Tak przygotowaną próbkę miareczkowano 0,1 M roztworem tiosiarczanu sodowego do chwili zmiany zabarwienia z brunatnoniebieskiego na białe.
Obliczenie zawartości garbników:
Obliczenie zawartości garbników w 50 ml ekstraktu wodnego:
X = C - (a - 3b) x 6,354 x 1,2518
gdzie:
X – ilość garbników (w mg) w 50 ml wyciągu wodnego,
a – ilość ml ściśle 0,1 M roztworu octanu miedziowego użytego do oznaczenia,
b – ilość ml ściśle 0,1 M roztworu Na2S2O3 użyta do zmiareczkowania miedzi niezwiązanej przez garbniki,
c – ciężar garbnikanów miedzi (w mg),
6,354 – ilość mg miedzi w 1 ml 0,1 M roztworu octanu miedziowego,
1,2518 – stosunek mas cząsteczkowych.
Obliczenie zawartości garbników w badanym surowcu (% wagowe):
Y = 2x/d
gdzie:
Y – zawartość garbników w badanym surowcu,
x – obliczona zawartość garbników (mg) w 50 ml wyciągu wodnego,
d – odważka surowca (g).
Wyniki ilościowego oznaczenia garbników przedstawiono w tabeli 12.
Tabela 12. Zawartość procentowa garbników w Harpagophyti radix wyznaczona według metody opisanej w FP IV (5).
| Numer próby | Masa garbnikanów miedzi (mg) | Ilość ml ściśle 0,1N roztworu
Na2S2O3 użyta do zmiareczkowania miedzi nie związanej przez garbniki | Zawartość procentowa garbników w surowcu (%) | Zawartość procentowa garbników w surowcu w przeliczeniu na suchą masę (%) |
I
II
III | 20,5
21,9
21,1 | 8,5
8,45
8,5 | 4,90
4,94
5,02 | 5,47
5,51
5,60 |
| Średnia zawartość garbników (%) | 4,95 | 5,53 |
Analiza TLC
Odważono dwie próbki po 2,0 g rozdrobnionego surowca i przeniesiono do kolb okrągłodennych. Jedną z nich zalano wodą, a drugą 4% roztworem kwasu siarkowego. Kolby umieszczono we wrzącej łaźni wodnej pod chłodnicami zwrotnymi i ogrzewano przez 3 godz. Następnie ekstrakty przesączono przez zwitki waty do rozdzielaczy i ekstrahowano octanem etylu. Ekstrakty octanowe osuszono bezwodnym siarczanem sodu i odparowano. Suchą pozostałość rozpuszczono w niewielkiej ilości metanolu i przeprowadzono analizę TLC wobec wzorców na płytkach pokrytych poliamidem w układach rozwijających:
metanol:woda:kwas octowy (20:12,5:1 v/v),
metanol:woda:dioksan:kwas octowy (20:12:1:1 v/v),
na płytkach pokrytych żelem krzemionkowym Si-60 G F254 w układzie:
toluen: mrówczan etylu: kwas mrówkowy (5:4:1 v/v)
oraz na płytkach pokrytych celulozą w układach rozwijajacych:
woda:dioksan (5:1 v/v),
woda:dioksan (10:1 v/v).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Wolski T, Baj T, Ludwiczuk A i wsp. Hakorośl rozesłana ( Harpahophytum procumbens DC.) – roślinny surowiec o wielokierunkowym działaniu farmakologicznym. Post Fitoter 2010; (1);13-22. 2. ESCOP Monographs. Thieme 2009. 3. Farmakopea Polska V, PTF, Warszawa 1999. 4. European Pharmacopoeia 3rd Edition. 1997. 5. Farmakopea Polska IV, T II, PZWL, Warszawa 1970. 6. Blumenthal M i wsp. The complete German Commission E Monographs – Therapic guide to herbal medicines. 1998; 248-50. 7. Bradley PR. British herbal compendium. 1992; 78-80. 8. Czygan F-C, Krüger A, Schier W i wsp. Pharmazeutisch-biologische Untersuchungen der Gattung Harpagophytum (Burch.) DC. ex Meissn. 1. Mitteilung: Phytochemische Standardisierung von Tubera Harpagophyti. Dtsch Apoth Ztg 1977; 117:1431-4. 9. Hojden B. Czarci pazur – ziele o działaniu przeciwreumatycznym. Wiad Ziel 1994; 36(11). 10. Lamer-Zarawska E. Lecznicze właściwosci Harpagophytum procumbens. Wiad Ziel 2000; 10. 11. Nervall CA, Anderson LA, Phillipson JD. Herbal Medicines, The Pharmaceutical Press 1996; 296. 12. Schmidt M, Eich J, Kreimeyer J i wsp. Teufels-kralle Sichere pharmaceutische Qualitat durch kontrollierten Anbau. Dtsch Apoth Ztg 1998; 47(138):4540-9. 13. Wegener T, Wiedenbrück R. Die Teufelskralle ( Harpagophytum procumbens DC.) in der Therapie rheumatischen Erkrankungen. Ztsch f Phytother 1998; 19:284-94. 14. www.emea.eu.int/pdfs/vet/mvls/067099en.pdf. 15. Czygan F-C. Harpagophytum – Teufelskralle. Ztsch f Phytother 1987; 8:17-20. 16. Farmakopea Niemiecka DAB 10. 1993. 17. Borkowski B, Miłkowska K. Garbniki, tanoidy i związki pokrewne IV. Kawolidy. Herba Pol 1996; 42(3):174-81.