© Borgis - Postępy Nauk Medycznych 3/2011, s. 222-231
*Urszula Wereszczyńska-Siemiątkowska1, Andrzej Siemiątkowski2
Mediatory procesu zapalnego i koagulopatii w ostrym zapaleniu trzustki – wybrane zagadnienia
Mediators of inflammatory and coagulation processes in acute pancreatitis – selected issues
1Klinika Gastroenterologii i Chorób Wewnętrznych Uniwersytetu Medycznego w Białymstoku
Kierownik Kliniki: prof. dr. hab. med. Andrzej Dąbrowski
2Klinika Anestezjologii i Intensywnej Terapii Uniwersytetu Medycznego w Białymstoku
Kierownik Kliniki: dr hab. med. Andrzej Siemiątkowski
Streszczenie
W ciężkim ostrym zapaleniu trzustki (OZT) miejscowa, jak i uogólniona reakcja zapalna jest wyrazem interakcji między aktywowanymi komórkami śródbłonkowymi a leukocytami, płytkami krwi i komórkami tucznymi oraz uwolnionymi z nich mediatorami. Narastająca nieadekwatna i niewłaściwie skierowana odpowiedź zapalna prowadzi w formie systemowego zapalenia (Systemic Inflammatory Response Syndrome, SIRS) i koagulopatii do rozwoju niewydolności narządowej w OZT. Ważną rolę odgrywają w tym procesie cytokiny prozapalne (TNFα, IL-1, IL-6, IL-8, IL-18), których aktywność prowadzi do przełamania kompensacyjnej odpowiedzi przeciwzapalnej (Compensatory Antiinflammatory Response Syndrome, CARS) związanej z potencjalnie przeciwzapalnymi cytokinami (IL-10, IL-2, IL-4, IL-1ra) oraz cząsteczki adhezyjne odpowiedzialne za selektywną rekrutację subpopulacji krążących leukocytów do miejsca zapalenia. Wczesne uruchomienie kaskady śródbłonkowych czynników krzepnięcia, szczególnie aktywacja czynnika tkankowego, jest integralną składową tego procesu. W niniejszym opracowaniu przedstawiono opublikowane wyniki badań własnych.
Summary
In the severe form of acute pancreatitis (AP) both, local and systemic inflammatory response reflects the interaction between activated endothelial cells (ECs) and leucocytes, thrombocytes, mast cells and released by them mediators. Increasing inadequate and not properly orientated inflammatory response, in the form of systemic inflammation (SIRS) and coagulopathy, leads to the development of multiorgan insufficiency in AP. A salient role in this process is attributed to proinflammatory cytokines (TNFα, IL-1, IL-6, IL-8, IL-18), whose activity leads to impairment of the compensatory antiinflammatory response syndrome (CARS), dependent of the antiinflammatory cytokines (IL-10, IL-2, IL-4, IL-1ra), and adhesive molecules accountable for selective recruitment of circulating leukocytes subpopulations to the inflammation site. The early commencement of endothelial coagulation factors cascade, especially tissue factor (TF) activation, is an integral component of this process. In this review the published results of the authors’ studies have been presented.

Ostre zapalenie trzustki (OZT) jest chorobą, która u progu XXI wieku, w jej ciężkiej postaci, nadal jest źródłem znaczącej chorobowości i śmiertelności. Całkowita śmiertelność w OZT wynosi 6-10%, wykazując tendencję zniżkową, co wiąże się z ustaleniem pewnych zasad postępowania, jednakże przy martwicy jałowej wzrasta do 10%, przy martwicy zakażonej do 25%, zaś współistnienie niewydolności narządowej zwiększa śmiertelność do 50% (1, 2, 3, 4).
W przebiegu ciężkiej postaci OZT występują dwa okresy zwiększonej śmiertelności (ryc. 1). Okres wczesny, tj. w ciągu pierwszych 7-10 dni od początku choroby, z około 60% śmiertelnością, spowodowaną wczesną i przetrwałą niewydolnością narządową, uwarunkowaną obecnością zespołu uogólnionej reakcji zapalnej (Systemic Inflammatory Response Syndrome, SIRS) i, jak ostatnio wykazano, bakteriemii, która stwierdzana była u 50% pacjentów. Okres późny, po upływie 3-4 tygodni od początku choroby, ze śmiertelnością sięgającą 40%, związany jest z różnie długim okresem supresji immunologicznej sprzyjającej rozwojowi powikłań infekcyjnych, sepsy z wtórną niewydolnością wielonarządową (2, 4, 5).
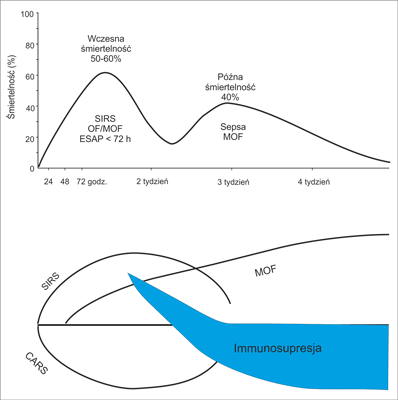
Ryc. 1. Dwa okresy zwiększonej śmiertelności w ostrym zapaleniu trzustki. ESAP – wczesne ciężkie ostre zapalenie trzustki (early severe acute pancreatitis), OF – niewydolność narządowa (organ failure), MOF – niewydolność wielonarządowa (multiorgan failure), SIRS – zespół uogólnionej reakcji zapalnej (systemic inflammatory response syndrome), CARS – zespół kompensacyjnej odpowiedzi przeciwzapalnej (compensatory antiinflammatory response syndrome).
Jednym z najistotniejszych ogniw patogenezy i rozwoju ostrego zapalenia trzustki jest proces interakcji między krążącymi komórkami krwi a śródbłonkiem naczyniowym zarówno miejscowo w trzustce, jak i systemowo w odległych narządach. Po raz pierwszy H. Rinderknecht (6) już 1988 roku zwrócił uwagę na znaczenie nadmiernej aktywacji leukocytów w patofizjologii OZT, rozpoczynając cykl badań poszerzających wiedzę o mediatorach odpowiedzi zapalnej i immunologicznej w tej chorobie. Pomostem w rozumieniu zależności pomiędzy leukocytami a komórkami śródbłonkowymi w OZT było sformułowanie przez Bone’a w 1992 roku pojęcia zespołu uogólnionej reakcji zapalnej (Systemic Inflammatory Response Syndrome, SIRS) (7). W zaproponowanej przez Bone’a trójstopniowo przebiegającej reakcji zapalnej, SIRS jest ostatnim etapem naturalnie wzbudzanej odpowiedzi organizmu na uszkodzenie, tj. po wyzwoleniu mediatorów miejscowo w fazie pierwszej, następnie przenikaniu ich w ograniczonych i kontrolowanych ilościach do krążenia w fazie drugiej, w trzeciej następuje przełamanie mechanizmów regulujących z masywnym wzbudzeniem i uwolnieniem mediatorów do krążenia (ryc. 2).
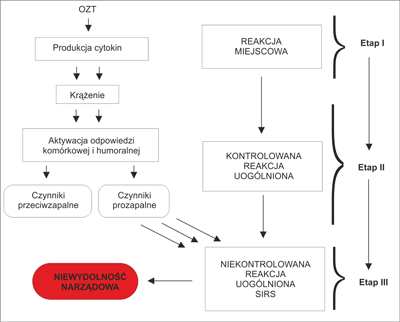
Ryc. 2. Schemat zespołu uogólnionej rekcji zapalnej (systemic inflammatory response syndrome).
W ostrym zapaleniu trzustki uszkodzenie komórki pęcherzykowej trzustki, związane z procesem przedwczesnej aktywacji proenzymów trzustkowych z dominującą rolą trypsyny i zaburzeniem spolaryzowanego procesu wydzielniczego, leży u podstawy patomechanizmu uszkodzenia i rozwoju martwicy miąższu trzustki oraz aktywacji i uwolnienia licznych mediatorów prozapalnych, m.in. jak reaktywnych postaci tlenu (RPT), czynnika aktywującego płytki krwi (Platelet Activating Factor, PAF), tlenku azotu (NO), leukotrienów (np. LTB4), układu humoralnego kinin, fibrynolizy, krzepnięcia i dopełniacza (8, 9, 10, 11, 12). W obecności mediatorów procesu zapalnego dochodzi do aktywacji bytujących makrofagów, komórek tucznych oraz do selektywnej rekrutacji subpopulacji leukocytów i nacieku podścieliska łącznotkankowego trzustki przez neutrofile, monocyty/makrofagi i limfocyty, które po aktywacji wraz z aktywowanymi komórkami śródbłonkowymi stają się kolejnym źródłem madiatorów nasilającym i podtrzymującym proces zapalny (12, 13, 14, 15). W wyniku masywnej reakcji immunologiczno-zapalnej w trzustce, w obecności cytokin prozapalnych (TNFα, IL-1, IL-6, IL-8, IL-18) dochodzi do przełamania kompensacyjnej odpowiedzi przeciwzapalnej (Compensatory Antiinflammatory Response Syndrome, CARS) związanej z uwalnianiem cytokin potencjalnie przeciwzapalnych: interleukina 10 (IL-10), interleukina 2 (IL-2), interleukina 4 (IL-4), antagonista receptora interleukiny 1 (IL-1ra) i przejścia miejscowej odpowiedzi zapalnej w trzustce w zapalenie ogólnoustrojowe (SIRS). Zwiększenie przepuszczalności i uszkodzenie śródbłonka naczyniowego jest morfologicznym wykładnikiem SIRS. Zatem narastająca, nieadekwatna i niewłaściwie skierowana odpowiedź zapalna prowadzi w formie systemowego zapalenia i koagulopatii do rozwoju niewydolności narządowej w OZT (ryc. 3) (9, 10, 15, 16, 17).
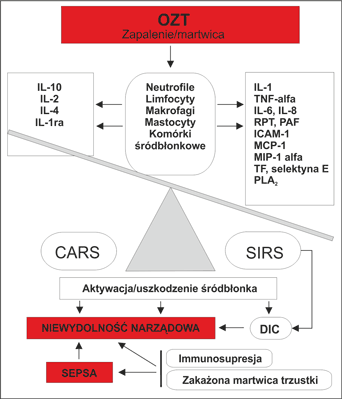
Ryc. 3. Patogeneza ostrego zapalenia trzustki.
SIRS – zespół uogólnionej reakcji zapalnej (systemic inflammatory response syndrome), CARS – zespół kompensacyjnej odpowiedzi przeciwzapalnej (compensatory antiinflammatory response syndrome).
Wyniki badań ostatniej dekady dotyczące patogenezy OZT w dużym stopniu ugruntowały już miejsce i znaczenie cytokin i cząsteczek adhezyjnych jako dynamicznego ogniwa w tym procesie. Natomiast coraz szersza jest wiedza dotycząca roli czynników krzepnięcia w rozwoju SIRS i koagulopatii w OZT. Większe zrozumienie zależności między tymi mediatorami umożliwiły badania wskazujące, iż w bardzo wczesnej fazie rozwoju OZT stwierdza się aktywację prozapalnych czynników transkrypcyjnych, tj. jądrowego czynnika transkrypcyjnego – kB (Nuclear Factor-kB, NFkB/Rel), białka aktywującego-1 (Activating Protein-1, AP-1), aktywowanych stresem kinaz, tj. kinazy aktywowanej mitogenami (Mitogen-Acivated Protein Kinase, MAPK), kinazy JUN (c-Jun Amino-Terminal Kinase). NF-kB jest pierwotnym regulatorem ekspresji genów wielu prozapalnych mediatorów, tj. cytokin (TNFα, IL-1, IL-8, IL-18), cząsteczek przylegania (selektyna E, ICAM-1) i czynnika tkankowego (TF) (15, 17, 18, 19, 20, 21).
Cytokiny i cząsteczki adhezyjne w ostrym zapaleniu trzustki
Cytokiny i cząsteczki adhezyjne są ważnymi modulatorami odpowiedzi immunologiczno-zapalnej zarówno we wczesnym, jak i późnym okresie OZT.
Spośród cytokin najwcześniej uwalnianymi mediatorami zapalenia w OZT są czynnik martwicy nowotworu (TNFα) i interleukina 1 (IL-1). TNFα i IL-1 działają wzajemnie synergistycznie, jak również z interleukiną 2 (IL-2) i interleukiną 6 (IL-6), oraz chemotaktycznie i aktywująco na neutrofile, monocyty i makrofagi. Biorąc pod uwagę aktywność biologiczną TNFα i IL-1 oraz fakt, iż są uwalnianie w OZT nie tylko w trzustce, ale również w wątrobie, płucach i śledzionie, cytokiny te zostały uznane głównymi czynnikami inicjującymi zarówno miejscową, jak i uogólnioną reakcję zapalną oraz ważnymi mediatorami rozwoju niewydolności wielonarządowej w tej chorobie (12, 15, 17).
IL-6 jest cytokiną prozapalną produkowaną przez fagocyty jednojądrowe i komórki śródbłonkowe w odpowiedzi na stymulację IL-1 i TNFα. Jest cytokiną dobrze scharakteryzowaną w OZT jako pierwotny czynnik wzbudzający produkcję białek ostrej fazy w wątrobie i wczesny wskaźnik prognostyczny stopnia ciężkości OZT. W badaniach własnych (ryc. 4) stwierdzaliśmy w pierwszych trzech dobach od chwili przyjęcia znamiennie najwyższe stężenie IL-6 w surowicy chorych z ciężką postacią OZT w odniesieniu do grupy kontrolnej, łagodnej postaci OZT oraz pacjentów z ostrymi nietrzustkowymi chorobami zapalnymi jamy brzusznej (OZNT). Równocześnie w przeprowadzonej analizie ROC w pierwszym dniu hospitalizacji wykazano, że IL-6 podobnie do selektyny E i elastazy neutrofilów ma wysoką zgodność prognostyczną w przepowiadaniu ciężkości OZT (9).
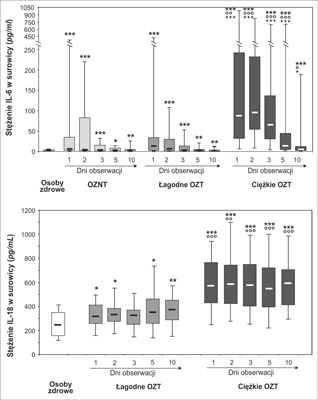
Ryc. 4. Stężenie interleukiny 6 (IL-6) w surowicy u pacjentów z łagodnym i ciężkim ostrym zapaleniem trzustki oraz w grupie pacjentów z OZNT, ostrymi zapalnymi nietrzustkowymi chorobami jamy brzusznej (9) i stężenie interleukiny 18 (IL-18) w surowicy u pacjentów z łagodnym i ciężkim ostrym zapaleniem trzustki (10).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Frossard J-L, Steer M, Pastor C M: Acute pancreatitis. Lancet 2008; 371: 143-52.
2. Fu Ch-Y, Yeh Ch-N, Hsu J-T et al.: Timing of mortality in severe acute pancreatitis: Experience from 643 patients. World J Gastroenterol 2007; 13: 1966-69.
3. Tonsi AF, Bacchion M, Crippa S et al.: Acute pancreatitis at the beginning of the 21st century: The state of the art. World J Gastroenterol 2009; 15: 2945-59.
4. Lankisch PG: Natural course of acute pancreatitis. Pancreas 2009; 38: 494-8.
5. Beger HG, Rau BM: Severe acute pancreatitis: Clinical course and management. World J Gastroenterol 2007; 14: 5043-51.
6. Rinderknecht H: Fatal pancreatitis: a consequence of excessive leukocyte stimulation? Int J Pancreatol 1988; 3: 105-112.
7. Bone RC, Balk RA, Cerra FB et al.: Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. ACCP/SCCM Consensus Conference Committee. American College of Chest Physicans/Society of Critical Care Medicine. Chest 1992; 101: 1644-1655.
8. Dąbrowski A, Gabryelewicz A: Oxidative stress – an early phenomenon characteristic of acute experimental pancreatitis. Int J Pancreatol 1992; 12: 193-199.
9. Wereszczyńska-Siemiątkowska U, Dąbrowski A, Siemiątkowski A et al.: Serum profiles of E-selectin, interleukin-10, interleukin-6 and oxidative stress parameters in patients with acute pancreatitis and non-pancreatic acute abdominal pain. Pancreas 2003; 26: 144-152.
10. Wereszczyńska-Siemiątkowska U, Mroczko B, Siemiatkowski A et al.: The importance of interleukin 18, glutathione peroxidase and selenium concentration changes in acute pancreatitis. Dig Dis Sci 2004; 49: 642-650.
11. Wereszczyńska-Siemiątkowska U, Długosz JW, Siemiątkowski A et al.: Lysosomal activity of pulmonary alveolar macrophages in acute experimental pancreatitis in rats with reference to positive PAF-antagonist (BN 52021) effect. Exp Toxic Pathol 2000; 52: 119-125.
12. Bhatia M, Wrong FL, Cao Y et al.: Pathophysiology of acute pancreatitis. Pancreatology 2005; 5: 132-144.
13. Pietruczuk M, Dąbrowska M I, Wereszczyńska-Siemiątkowska U, Dąbrowski A: Alteration of peripheral blood lymphocyte subsets in acute pancreatitis. World J Gastroenterol 2006; 12: 5344-51.
14. Dąbrowski A, Osada J, Dąbrowska MI, Wereszczyńska-Siemiątkowska U: Monocyte subsets and natural killer cells In acute pancreatitis. Pancreatology 2008; 8: 126-34.
15. Wereszczyńska-Siemiątkowska U, Siemiątkowski A: Rola układu immunologicznego w ostrym zapaleniu trzustki – znaczenie cytokin i cząsteczek przylegania. Med Sci Rev 2002; 1: 84-90.
16. Wereszczynska-Siemiatkowska U, Mroczko B, Siemiatkowski A: Serum profiles of interleukin-18 in different severity forms of human acute pancreatitis. Scand J Gastroenterol 2002; 37: 1097-1102.
17. Wereszczyńska-Siemiątkowska U, Kamiński K: Rola cytokin i cząsteczek przylegania w ostrym zapaleniu trzustki. Gastroenterol Pol 2000; 7: 319-323.
18. Dąbrowski A, Tribiłło I, Dąbrowska MI et al.: Activation of mitogen-activated protein kinases In different models of pancreatic acinar cell damage. Z Gastroenterol 2000; 38: 469-81.
19. Guzman EA, Rudnicki M: Intricacies of host response in acute pancreatitis. Am College Surg 2006; 202: 509-19.
20. Kakafika A, Papadopoulos V, Mimidis K, Mikhailidis DP: Coagulation, platelets and acute pancreatitis. Pancreas 2007; 34: 15-20.
21. Wereszczyńska-Siemiątkowska U, Kosel J, Siemiatkowski A: Właściwości biologiczne interleukiny 18. Pol Merk Lek 2004; XVI, 93: 279-81.
22. Rau B, Baumgart K, Paszkowski AS et al.: Clinical relevance of caspase-1 activated cytokines in acute pancreatitis: high correlation of serum interleukin-18 with pancreatic necrosis and systemic complications. Crit Care Med 2001; 29: 1556-62.
23. Paszkowski AS, Rau B, Mayer JM et al.: Theraputic application of caspase 1/interleukin-1β-converting enzyme inhibitor decreases the death rate in severe acute experimental pancreatitis. Ann Surg 2002; 235: 68-76.
24. Oberholzer A, Feilner A, Hentze H et al.: Sepsis after severe injury interrupts caspase-dependent processing of interleukin-18. J Trauma 2000; 49: 11-17.
25. Daniel P, Leśniowski B, Jasińska A et al.: Usefulness of assessing circulating levels of resistin, ghrelin, and IL-18 in alcoholic acute pancreatitis. Dig Dis Sci 2010; 55: 2982-7.
26. Wereszczyńska-Siemiątkowska U, Siemiątkowski A, Mroczko B et al.: Serum interleukin 2 (IL-2) and its soluble receptor (sIL-2R) concentrations In patients with severe form of acute pancreatitis (AP). GUT 2003; 52 (Suppl VI) A9.
27. Pezzilli R, Corsi M, Barassi A et al.: Serum adhesion molecules in acute pancreatitis. Pancreas 2008; 37: 36-41.
28. Siemiątkowski A, Wereszczyńska-Siemiątkowska U, Borkowski J, Dabrowski A: Coagulation factors and adhesion molecules of endothelial origin in the different severity forms of acute pancreatitis. Pancreatology 2004; 4 (Suppl): 157.
29. Levi M, Cate H, Van der Poll T: Endothelium: interface between coagulation and inflammation. Crit Care Med 2002; 30 (Suppl): 20-224.
30. Doshi SN, Marmur JD: Evolving role of tissue factor and its pathway inhibitor. Crit Care Med 2002; 30: 241-250.
31. Sawa H, Ueda T, Takeyama Y et al.: Elevation of plasma tissue factor levels in patients with severe acute pancreatitis. J Gastroenterol 2006; 41: 575-81.
32. Yasuda T, Ueda T, Kamei K et al.: Plasma tissue factor pathway inhibitor levels in patients with acute pancreatitis. J Gastroenterol 2009; 44: 1071-79.