© Borgis - Postępy Nauk Medycznych 4/2011, s. 344-351
*Ryszard Gellert
Nefropatia cukrzycowa 2010 – zapowiedź zmian w praktyce klinicznej?
Diabetic nephropathy 2010 – an omen of change in clinical practice?
Department of Nephrology Medical Center of Postgraduate Education
Head of Department: prof. dr hab. med. Ryszard Gellert
Streszczenie
Opublikowane w ostatnim czasie dane dotyczące patogenezy zmian kłębuszkowych w cukrzycy oraz wyniki kontrolowanych dużych badań klinicznych wskazują na istotną rolę optymalnej kontroli glikemii dla prewencji nefropatii cukrzycowej. Hiperglikemia prowadzi bowiem do zwiększenia ekspresji ACE w endotelium naczyń kłębuszkowych i zmniejsza aktywność ACE2 na podocytach, co ma prowadzić do albuminurii. Hamowanie układu renina angiotensyna we wczesnych etapach nefropatii cukrzycowej zmniejsza albuminurię ale nie spowalnia progresji choroby. Hiperfiltracja we wczesnych stadiach cukrzycy nie jest najprawdopodobniej przyczyną białkomoczu. Zmniejszone unieczynnianie angiotensyny II przez ACE2 ma też prowadzić do uszkadzania struktury podocytów i ich utraty do moczu, co ma skutkować szkliwieniem kłębuszków. Zatem albuminuria i niewydolność nerek nie są ze sobą powiązane i traktowanie albuminurii jako markera nefropatii cukrzycowej i skuteczności nefroprotekcji nie ma uzasadnienia. W późniejszych stadiach nefropatii cukrzycowej hamowanie układu reniana-angiotensyna jest skuteczne tylko przy współistnieniu nadciśnienia tętniczego. Dlatego zalecane dotychczas rutynowe stosowanie ACEi i/lub ARB w hamowaniu jej progresji musi być ograniczone do pacjentów z nadciśnieniem tętniczym. Utrzymane natomiast muszą być zalecenia co do hamowania układu renina-angiotensyna w prewencji makroangiopatii. Kontrola glikemii i nadciśnienia tętniczego mają kluczowe znaczenie dla nefroprotekcji w cukrzycy. Celem leczenia jest uzyskanie odsetka HbA1c < 7,0%, także w DM2, i normalizacji ciśnienia tętniczego. W zaawansowanej nefropatii cukrzycowej szczególnie przydatne dla nefroprotekcji są inhibitory ACE, które w przypadku nietolerancji można zastąpić sartanami. Nadzieję na poprawę nefroprotekcji w cukrzycy rokują opracowywane ACE2mimetyki.
Summary
Recently published data on the pathogenesis of glomerular changes in diabetes, and on the outcomes of big randomised clinical trials, stress the importance of strict glycaemia control in preventing diabetic nephropathy. Hyperfiltration does not cause albuminuria in the early phase of diabetic nephropathy. Hyperglycaemia results in over-expression of ACE on the glomerular endothelium and decreases the ACE2 activity on podocytes, which increases angiotensin II and initiates albuminuria. Inhibition of renin-angiotensin system does not prevent diabetic nephropathy, but decreases albuminuria in the early phase of diabetic nephropathy without impeding the progression of the disease. In more advanced stages of diabetic nephropathy inhibition of renin-angiotensin system impedes the progression of renal failure only in hypertensive patients. The decreased inactivation of increased amount of angiotensin II by ACE2 harm the podocyte skeleton, second to the augmented local angiotensin II activity. This results in shedding podocytes into urine /podocyturia/, which uncovers the basal membrane and initiates glomerular hyalinisation and sclerosis. Thus, albuminuria and renal failure are not causally linked to each other, and there is no ground for interpreting albuminuria as a marker of diabetic nephropathy, nor to consider changes in albuminuria a prognostic marker of nephroprotection. Prescribing ACEi and/or ARBs as nephroprotection to every patient with diabetes, which is a routine today, should be limited to patients with coexisting hypertension and albuminuria. The indication to prescribe ACEi/ARBs in preventing macroangiopathy is kept. Controlling hypertension is important for nephroprotection along with controlling glycaemia. The target of therapy is to normalize blood pressure and to keep A1C < 7.0%, also in diabetes type 2. In advanced nephropathy ACEi are especially useful and, in case of intolerance should be replaced with ARBs. The ACE2-mimetics, which are under development, raise hope for further improvement of nephroprotection in diabetes.

In the last decade diabetic nephropathy (DN) became a more and more frequent reason for starting chronic renal replacement therapy – in Poland, in 2007 DN affected 25.3% of incident dialysis patients (1). The incidence of dialysis consequent to DN positively correlates with the prevalence of diabetes in general population (2). Type 2 diabetes (DM2) is more prevalent in dialysis patients as compared to diabetes type 1 (DM1). The only procedure to establish the definite diagnosis of DN is renal biopsy. This procedure can be abandoned, if feasible for technical reasons, only in patients presenting with isolated albuminuria, long-lasting diabetes (over 5 years in DM1, and 10 years in DM2), diabetic retinopathy and slowly deteriorating renal function. Lack of any of the four aforementioned conditions should alert the physician to consider the (co)existence of non-diabetic glomerulopathy, including the crescentic glomerulonephritis.
ALBUMINURIA AND PROTEINURIA FOR DIAGNOSING DN
Contrary to the common opinion, albuminuria is not an early marker of DN (3), for it can present in advanced DN only, or never. Exactly for the same reason, albuminuria is not a good marker of advanced DN, as well. Even the proteinuria accompanying extrarenal diabetic microangiopathy is not a proof of DN – the diabetic retinopathy was present in 10% of DM2 patients with biopsy-proven non-diabetic glomerulopathy (4). Moreover, retinopathy was present only in 70.8% (4)-72% (5) DM2 patients with biopsy-proven DN. However, in DM1 patients presenting DN the retinopathy is usually present.
ALBUMINURIA AND PROTEINURIA IN DN PROGRESSION
Proteinuria, regardless of its range – from microalbuminuria to nephrotic syndrome – in diabetic patients is, like in general population, an universally accepted morbidity and cardiovascular (macroangiopathy) and renal (microangiopathy) mortality risk factor. However, the diagnostic significance of albuminuria seems to change in the course of disease.
Proteinuria was proven to damage renal tubules and promote renal interstitial fibrosis (6, 7), but cannot be considered the exclusive reason of renal filtration decrease in DN. Many patients with membranous (MGN) or minimal change glomerulopathy (MCD) do not progress to renal failure, despite the massive, perennial proteinuria. Also in experimental conditions, the massive proteinuria, second to damaging the slit diaphragm with antinephrin antibodies does not result in renal interstitial fibrosis and renal failure (8). On the other hand, smoking cessation 35 and body mass reduction (9), factors which do not affect glomerular structure directly, diminish proteinuria and decelerate glomerular filtration loss (nephroprotection). Thus, the proteinuria per se should not be considered the only nor the sufficient condition of nephropathy progression (10, 11).
The response to hypotensive therapy meta-analysed in 33 clinical trials (77 therapeutic groups) unambiguously revealed, that the diminishing the proteinuria correlated with the retardation of renal failure progression only in advanced DN (DM1, r = -0.67, p = 0.03; DM2, r = -0.57, p = 0.02). Such a correlation could not be observed at early stages of DM1, normotensive DM2, nor hypertensive DM2 (12). Contrary to what is observed in advanced DN, the decrease in albuminuria second to pharmacological intervention was not a predictor of efficient nephroprotection at early DN (12).
The above observations suggest that in diabetic patients proteinuria and loss of glomerular filtration could reflect different disease processes, not necessarily of distinct aetiology. For the renal failure appears to develop and progress independently from albuminuria, at least at early stage of DN, neither the albuminuria should be considered a predictor of renal failure, nor changes in albuminuria should in DN be used as an index of risk in renal function changes.
The clinical observations mentioned above got lately a further reinforcement from the experimental research on the pathophysiology of diabetic glomerulopathy, which gave a well documented rationale to separate the pathogenesis of albuminuria from that of renal failure progression.
PATHOGENESIS OF ALBUMINURIA IN DIABETIC NEPHROPATHY
Typical diabetic changes in kidneys are present mainly in glomeruli. These are mesangial cell proliferation and mesangial matrix expansion, thickening of the glomerular basement membrane, loss of podocytes, and changes in their morphology –foot processes effacement, and finally the hyalinisation and sclerotisation of glomerular capillaries. These changes augment, in DM1 and DM2, along with the increase of proteinuria (13, 14) secondary to the damage to both, the glomerular filtration barrier and to the impairment of podocyte cytoskeleton.
In early DN, hyperfiltration is very common. Contrary to the universal belief, the hyperfiltration does not seem to cause albuminuria – after 15 years of follow-up in DM1 patients, the prevalence of proteinuria in presenting hyperfiltration at inclusion, did not differ as compared to these with normal filtration at the beginning of observation (19% and 23%, respectively) (15).
The basic indicator of diabetic damage to the glomerular filtration barrier is the decreased content of nephrin in the slit membrane (16, 17). The content of other proteins linked to nephrin -podocin and podocalixin, remains unchanged in DN (18). The decreased activity of nephrin appears to result from the over-activity of angiotensin II (AngII), for the inhibition of the angiotensin system restores nephrin expression under experimental conditions (19). The increased AngII activity appears to be of particular importance to damaging the podocyte cytoskeleton (20), second to activation of the protein kinase C (PKC). The last one is hold responsible also for mesangial proliferation, which occurs along with the inhibition of apoptosis, secondary to the hyperglycaemia-induced decrease in p21 activity (21). It can be clearly seen, from the above, that AngII is of fundamental importance to changes in glomerular morphology and function, even if other factors, like the increased catepsin L (CatL) activity (22), are also involved.
The increased glomerular AngII activity results from both, hyperglycaemia and local deregulation of the ACE and ACE2 activity. The ACE2, first described in 2000, converts both, angiotensin I (AngI) into inactive angiotensin 1-9 (Ang1-9), and AngII into angiotensin 1-7 (Ang 1-7). The last acts antagonistically to AngII, what is schematically shown on Figure 1.
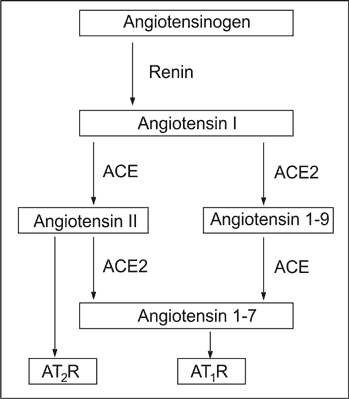
Fig. 1. The simplified Renin-Angiotensin-Aldosterone axis in renal glomerulus.
Within the glomerular tuft the ACE is localised mainly, if not exclusively, on the endothelial cells, and ACE2 can be identified exclusively on the podocyte surface23. The diagram below illustrates the biochemical consequences of such space-separation of ACE from ACE2.
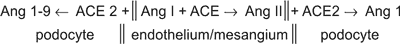
The inhibition ACE2, under experimental conditions, results in proteinuria, which is absent if AT1 receptors are blocked (23). The opposite effect – diminished proteinuria, can be seen after recombinant human ACE2 (rhACE2) is given to the diabetic mice (24).
In DM the glomerular ACE2 activity is diminished, which along with the increased ACE activity, can result in local AngII over-production, followed by slit diaphragm dysfunction/damage (23) and consequent albuminuria. Thus, one can conclude, the key factor in pathogenesis of diabetic proteinuria is a damage to the podocyte slit diaphragm resulting from the increased Ang II activity secondary to hyperglycaemia.
PATHOGENESIS OF RENAL FAILURE IN DIABETIC NEPHROPATHY
Urine of diabetic patients contains increased amount of podocytes, as compared to healthy controls. The increase in podocyturia is proportional to proteinuria. This is supposed to discriminate renal failure of diabetic origin from that caused by any other glomerulopathy (25). The inhibition of RAA activity with trandolapril normalised podocyturia both, in diabetic patients presenting micro- and macroalbuminuria (19). Thus, one can stipulate, with high level of probability, that diabetic glomerular damage results from the loss of podocytes caused by the increased AngII activity, not from proteinuria itself.
Hyperfiltration, podocyte effacement, and the dysfunction of the slit diaphragm seem to be fully reversible in early DN. Most probably the loss of numerous podocytes, secondary to prolonged or massive podocyturia, lays bare the basement membrane and results in irreversible glomerular hyalinisation and sclerotisation and consequent renal failure (26). The glomerular hyalinisation reduces the glomerular filtration area which results in renal failure, which can be further augment by the protein-induced tubular damage resulting in renal interstitial fibrosis.
ANGIOTENSIN II – THE COMMON ETIOLOGIC FACTOR OF DIABETES-INDUCED PROTEINURIA AND RENAL FAILURE
The pathophysiological data presented above, strongly suggest that early DN proteinuria results from the increased AngII activity, which impacts podocytes and results in a reversible dysfunction of slit diaphragm. The prolonged, or extremely intensive, AngII activity can also cause podocyte shedding, which results in reduction of area covered by podocytes on glomerular capillaries. This might explain, why there is no direct interrelationship between albuminuria/proteinuria and glomerular filtration loss in early DN. However, both phenomena are consequent of locally increased AngII activity. This, in turn, could explain why some diabetic patients present renal failure without albuminuria/proteinuria and some do not present renal failure despite long-lasting increased albumin excretion. The clinical practice clearly suggests the above reasoning could be meaningful.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Rutkowski B, Lichodziejewska-Niemierko M, Grenda R et al.: Raport o stanie leczenia nerkozastępczego w Polsce – 2007. Polski Rejestr Nefrologiczny. Drukonsul. Gdańsk 2009.
2. U.S. Renal Data System, USRDS 2008 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases, Bethesda, MD 2008.
3. Caramori ML, Kim Y, Huang C et al.:Cellular basis of diabetic nephropathy: 1. Study design and renal structural-functional relationships in patients with long-standing type 1 diabetes. Diabetes 2002; 51: 506-513. [Errata, Diabetes 2002; 51: 1294].
4. Wong TY, Choi PC, Szeto CC et al.: Renal outcome in type 2 diabetic patients with or without coexisting nondiabetic nephropathies. Diabetes Care 2002; 25: 900-5.
5. Christensen PK, Larsen S, Horn T et al.: Renal function and structure in albuminuric type 2 diabetic patients without retinopathy Nephrol Dial Transplant 2001; 6: 2337-47.
6. Simonson MS: Phenotypic transitions and fibrosis in diabetic nephropathy. Kidney Int 2007; 71: 846-54.
7. Noh H, King GL: The role of protein kinase C activation in diabetic nephropathy. Kidney International 2007; 72: S49-S53.
8. Kikuchi H, Kawachi H, Ito Y et al.: Severe proteinuria, sustained for 6 months, induces tubular epithelial cell injury and cell infiltration in rats but not progressive interstitial fibrosis. Nephrol Dial Transplant 2000; 15: 799-810.
9. Morales E, Valero MA, Leon M et al.: Beneficial effects of weight loss in overweight patients with chronic proteinuric nephropathies Am J Kidney Dis 2003; 41 (2): 319-27.
10. Kriz W, LeHir M: Pathways to nephron loss starting from glomerular diseases-insights from animal models. Kidney Int 2005; 67: 404-419 .
11. Gross ML, Hanke W, Koch A et al.: Intraperitoneal protein injection in the axolotl: the amphibian kidney as a novel model to study tubulointerstitial activation. Kidney Int 2002; 62: 51-59.
12. Jerums G, Panagiotopoulos S, Premaratne E et al.: Lowering of proteinuria in response to antihypertensive therapy predicts improved renal function in late but not in early diabetic nephropathy: a pooled analysis. Am J Nephrol 2008; 28: 614-627.
13. Toyoda M, Najafian B, Kim Y et al.: Podocyte Detachment and Reduced Glomerular Capillary Endothelial Fenestration in Human Type 1 Diabetic Nephropathy. Diabetes 2007; 56: 2155-2160.
14. Zhu WW, Chen H, Ge Y et al.: Ultrastructural changes in the glomerular filtration barrier and occurrence of proteinuria in Chinese patients with type 2 diabetic nephropathy, Diabetes Research and Clinical Practice 2009; 86: 199-207.
15. Ficociello LH, Perkins BA, Roshan B et al.: Renal hyperfiltration and the development of microalbuminuria in type 1 diabetes. Diabetes Care 2009; 32: 889-93.
16. Langham RG, Kelly DJ, Cox AJ et al.: Proteinuria and the expression of the podocyte slit diaphragm protein, nephrin, in diabetic nephropathy: effects of angiotensin converting enzyme inhibition. Diabetologia 2002; 45: 1572-6.
17. Doublier S, Salvidio G, Lupia E et al.: Nephrin expression is reduced in human diabetic nephropathy: evidence for a distinct role for glycated albumin and angiotensin II. Diabetes 2003; 52: 1023-30.
18. Koop K, Eikmans M, Baelde HJ et al.: Expression of podocyte-associated molecules in acquired human kidney diseases. J Am Soc Nephrol 2003; 14: 2063-71.
19. Wolf G, Chen S, Ziyadeh FN: From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes 2005; 54: 1626-1634.
20. Macconi D, Abbate M, Morigi M et al.: Permselective dysfunction of podocyte-podocyte contact upon angiotensin II unravels the molecular target for renoprotective intervention. Am J Pathol 2006; 168: 1073-85.
21. Danesh FR, Sadeghi MM, Amro N et al.: 3-Hydroxy-3-methylglutaryl CoA reductase inhibitors prevent high glucose-induced proliferation of mesangial cells via modulation of Rho GTPase/p21 signaling pathway: Implications for diabetic nephropathy. Proc Natl Acad Sci USA 2002; 99: 8301-5.
22. Sever S, Altintas MM, Nankoe SR et al.: Proteolytic processing of dynamin by cytoplasmic cathepsin L is a mechanism for proteinuric kidney disease. J Clin Invest 2007 Aug; 117 (8): 2095-104.
23. Ye M, Wysocki J, William J et al.: Glomerular localization and expression of Angiotensin-converting enzyme 2 and Angiotensin-converting enzyme: implications for albuminuria in diabetes. J Am Soc Nephrol 2006; 17: 3067-75.
24. Oudit GY, Liu GC, Zhong JiuChang et al.: Human Recombinant ACE2 Reduces the Progression of Diabetic Nephropathy. Diabetes 2010 Feb; 59 (2): 529-38.
25. Nakamura T, Ushiyama C, Suzuki S et al.: Urinary excretion of podocytes in patients with diabetic nephropathy. Nephrol Dial Transplant 2000; 15: 1379-83.
26. Mundel P, Reiser J: Proteinuria: an enzymatic disease of the podocyte? Kidney International 2010; 77: 571-580.
27. Mann F, Schmieder RE, Dyal L et al.: TRANSCEND (Telmisartan Randomised Assessment Study in ACE Intolerant Subjects with Cardiovascular Disease) Investigators. Effect of telmisartan on renal outcomes: a randomized trial Ann Intern Med 2009; 151: 1-10.
28. Bilous R, Chaturvedi N, Sj?lie AK et al.: Effect of candesartan on microalbuminuria and albumin excretion rate in diabetes: three randomized trials. Ann Intern Med 2009; 151 (1): 11-20.
29. Comper WD, Osicka TM, Jerums G: High prevalence of immuno-unreactive intact albumin in urine of diabetic patients. Am J Kidney Dis 2003; 41: 336-342.
30. Brinkman JW, Bakker SJ, Gansevoort RT et al.: Which method for quantifying urinary albumin excretion gives what outcome? A comparison of immunonephelometry with HPLC. Kidney Int Suppl 2004: S69-75.
31. Comper WD, Osicka TM, Clark M et al.: Earlier detection of microalbuminuria in diabetic patients using a new urinary albumin assay. Kidney Int 2004; 65: 1850-1855.
32. Comper WD, Osicka TM: Detection of urinary albumin. Adv Chronic Kidney Dis 2005; 12: 170-176.
33. Czekalski S, Grzeszczak W, Ciechanowski K, Renke M: Rozpoznawanie i leczenie nefropatii cukrzycowej. [W:] Rutkowski B, Czekalski S (red.): Standardy postępowania w rozpoznawaniu i leczeniu chorób nerek. Wydawnictwo Medyczne MAKmed, Gdańsk 2001.
34. Sawicki PT, Didjurgeit U, Mühlhauser I et al.: Smoking is associated with progression of diabetic nephropathy. Diabetes Care 1994; 17: 126-31
35. Chase HP, Garg SK, Marshall G et al.: Cigarette smoking increases the risk of albuminuria among subjects with type I diabetes. JAMA 1991; 265: 614-7.
36. Fouque D, Laville M: Low protein diets for chronic kidney disease in non diabetic adults. Cochrane Database Syst Rev 2009; Issue 3.
37. Pan Y, Guo LL, Jin HM: Low-protein diet for diabetic nephropathy: a meta-analysis of randomized controlled trials. Am J Clin Nutr 2008; 88: 660-666.
38. Parving H-H, Persson F, Lewis JB et al.: Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med 2008; 358: 2433-2446.
39. Lewis EJ, Hunsicker LG, Bain RP, Rohde RD: The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N Engl J Med 1993; 329: 1456-1462. [Errata, N Engl J Med 1993; 330: 152] .
40. Lewis EJ, Hunsicker LG, Clarke WR et al.: Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345: 851-860.
41. Brenner BM, Cooper ME, de Zeeuw D et al.: RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345: 861-869.
42. Gaede P, Vedel P, Larsen N et al.: Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med 2003; 348: 383-393.
43. DCCT Research Group: The association between glycemic exposure and longterm diabetic complications in the Diabetes Control and Complications Trial. Diabetes 1995; 44: 968-983,
44. Stratton IM, Adler AI, Neil HA et al.: Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 2000; 321: 405-412.
45. Epidemiology of Diabetes Interventions and Complications Research Group: Intensive diabetes therapy and carotid intima-media thickness in type 1 diabetes. N Engl J Med 2003; 48: 2294-2303.
46. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group: intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med 2005; 353: 2643-2653.
47. The Action to Control Cardiovascular Risk in Diabetes Study Group: Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med 2008; 358: 2545-2559.
48. The ADVANCE Collaborative Group: Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med 2008; 358: 2560-2572.
49. Abraira C, Duckworth WC, Moritz T: Glycaemic separation and risk factor control in the Veterans Affairs Diabetes Trial: an interim report. Diabetes Obes Metab. 29 July 2008 [Epub ahead of print].
50. Kausik K Ray: Sreenivasa Rao Kondapally Seshasai, Shanelle Wijesuriya, Rupa Sivakumaran, Sarah Nethercott, David Preiss, Sebhat Erqou, Naveed Sattar. Effect of intensive control of glucose on cardiovascular outcomes and death in patients with diabetes mellitus: a meta-analysis of randomised controlled trial. The Lancet 2009; 373: 1765-1772.
51. Diabetes Control and Complications Trial Research Group: The effect of intensive diabetes treatment on the development and progression of long-term complications in insulin-dependent diabetes mellitus: the Diabetes Control and Complications Trial. N Engl J Med 1993; 329: 978-986.
52. Reichard P, Nilsson B-Y, Rosenqvist U: The effect of long-term intensified insulin treatment on the development of microvascular complications of diabetes mellitus. N Engl J Med 1993; 329: 304-309.
53. UK Prospective Diabetes Study (UKPDS) Group: Intensive blood glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complication in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352: 837-853.
54. UK Prospective Diabetes Study (UKPDS) Group: Effect of intensive blood glucose control with metformin on complication in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998; 352: 854-865.
55. Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with NIDDM: a randomized prospective 6-year study. Diabetes Res Clin Pract 1995; 28: 103-117.
56. Skyler JS, Bergenstal R, Bonow RO et al.: Intensive Glycemic Control and the Prevention of Cardiovascular Events: Implications of the ACCORD, ADVANCE, and VA Diabetes Trials. A position statement of the American Diabetes Association and a scientific statement of the American College of Cardiology Foundation and the American Heart Association; Diabetes Care, 200932.
57. American Diabetes Association: Standards of medical care in diabetes-2008 (Position Statement). Diabetes Care 2008; 31 (Suppl 1): S12-S54.
58. Little RR, Rohlfing CL, Wiedmeyer H-M et al.: The National Glycohemoglobin Standardization Program (NGSP): a five year progress report. Clin Chem 2001; 47: 1985-1992.
59. Nathan DM, Buse JB, Davidson MB et al.: Medical Management of Hyperglycemia in Type 2 Diabetes: A Consensus Algorithm for the Initiation and Adjustment of Therapy. A consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes; Diabetes Care 2009; 32: 193-203.
60. http://www.kidney.org/professionals/KDOQI/guideline_diabetes/guide2.htm w dniu 28.09.2010
61. American Diabetes Association. Standards Of Medical Care In Diabetes-2009. Position Statement. Diabetes Care 2009; 32 (Suppl 1).