© Borgis - Postępy Nauk Medycznych 12/2011, s. 1008-1014
*Lidia Ziółkowska, Anna Turska-Kmieć, Agnieszka Boruc, Jadwiga Daszkowska-York, Maria Biernatowicz, Dorota Sobielarska-Łysiak, Monika Kowalczyk, Katarzyna Bieganowska, Wanda Kawalec
Obraz kliniczny kardiomiopatii przerostowej u dzieci – doświadczenia własne
Clinical profile of hypertrophic cardiomyopathy in children – own experience
Klinika Kardiologii Instytut „Pomnik – Centrum Zdrowia Dziecka”, Warszawa
Kierownik Kliniki: prof. dr hab. med. Wanda Kawalec
Streszczenie
Wstęp. Kardiomiopatia przerostowa jest genetycznie uwarunkowaną chorobą mięśnia sercowego, charakteryzującą się przerostem mięśnia lewej komory, który nie jest wtórny do wrodzonej wady serca lub nadciśnienia tętniczego. Jest chorobą o różnorodnym przebiegu klinicznym, często doprowadzającym do rozwoju niewydolności serca lub nagłego zgonu sercowego.
Cel pracy. Celem pracy była retrospektywna analiza obrazu klinicznego u dzieci z kardiomiopatią przerostową hospitalizowanych w Klinice Kardiologii Instytutu „Pomnik – Centrum Zdrowia Dziecka”.
Materiał i metody. Analizowano 85 pacjentów, w wieku średnio 10,7 lat, hospitalizowanych w latach 1991-2010.
Wyniki. Średni wiek pacjentów w chwili rozpoznania kardiomiopatii przerostowej wynosił 5,9 lat. Dodatni wywiad rodzinny w kierunku kardiomiopatii przerostowej stwierdzono u 41 (51,3%) dzieci. Nagłe zgony sercowe w rodzinie występowały u 24 (28,6%) dzieci. Pacjenci najczęściej zgłaszali męczliwość, upośledzenie tolerancji wysiłku (67,1%). Asymetryczny przerost mięśnia lewej komory stwierdzono u 53 (62,4%) pacjentów. U 11 (12,9%) dzieci wszczepiono kardiowerter-defibrylator, u 3 (3,5%) pacjentów w celu prewencji wtórnej oraz u 8 (9,4%) jako prewencję pierwotną nagłego zgonu sercowego. U 3 (3,5%) dzieci wystąpił nagły zgon sercowy, 6 (7,1%) pacjentów zmarło z powodu postępującej niewydolności serca.
Wnioski. W analizowanej grupie pacjentów stwierdzono bardziej obciążony wywiad rodzinny w kierunku występowania kardiomiopatii przerostowej i nagłych zgonów sercowych w rodzinie w porównaniu do wyników innych autorów. Najczęstszym objawem klinicznym była męczliwość, postępujące ograniczenie tolerancji wysiłku, epizody bólu w klatce piersiowej oraz omdlenia. U badanych dzieci stwierdzono większy przerost mięśnia sercowego w porównaniu z wynikami innych autorów. Częstość implantacji kardiowertera-defibrylatora w celu profilaktyki pierwotnej nagłego zgonu sercowego jest porównywalna z danymi piśmiennictwa.
Summary
Introduction. Hypertrophic cardiomyopathy is a genetically determined myocardial disease characterized by left ventricular hypertrophy, which is not secondary to congenital heart disease or hypertension. Is a disease with different clinical course, often is the cause of heart failure or sudden cardiac death.
Aim of study. Retrospective analysis of clinical profile in children with hypertrophic cardiomyopathy hospitalized in the Department of Paediatrc Cardiology.
Material and methods. We analyzed 85 patients, mean age 10.7 yrs hospitalized in the years 1991-2010.
Results. The mean age at diagnosis of hypertrophic cardiomyopathy was 5.9 years. A positive family history of hypertrophic cardiomyopathy was diagnosed in 41 (51.3%) children. Sudden cardiac death in the family occurred in 24 (28.6%) children. Patients most frequently reported fatigue, impaired exercise tolerance (67.1%). Asymmetric left ventricular hypertrophy was found in 53 (62.4%) patients. In 11 (12.9%) children cardioverter defibrillator was implanted, in 3 (3.5%) patients for secondary prevention and in 8 (9.4%) as primary prevention of sudden cardiac death. In 3 (3.5%) children sudden cardiac death occured, 6 (7.1%) patients died due to progressive heart failure.
Conclusions. In our group of patients were charged more family history of hypertrophic cardiomyopathy and sudden cardiac death. The most common clinical symptom was fatigue, a progressive reduction in exercise tolerance, episodes of chest pain and syncope. In the children tested were greater cardiac hypertrophy compared with the results of other authors. The frequency of implantable cardioverter-defibrillator for primary prevention of sudden cardiac death is comparable with the data of the literature.

Wstęp
Kardiomiopatia przerostowa (Hypertrophic Cardiomyopathy – HCM) jest chorobą mięśnia sercowego charakteryzującą się przerostem lewej komory. Klinicznie rozpoznawana jest u dzieci z przerostem mięśnia komory lewej, który nie jest wtórny do wrodzonej wady serca lub nadciśnienia tętniczego. Jest chorobą występującą od urodzenia, jakkolwiek przerost mięśnia lewej komory może być obecny od razu po urodzeniu lub we wczesnym okresie życia, ale w większości przypadków zmiany ujawniają się dopiero w okresie dojrzewania, po 12. roku życia. Stopień przerostu mięśnia sercowego, jak również etiologia kardiomiopatii przerostowej u dzieci jest różna, co wpływa na dużą różnorodność obrazu klinicznego i ekspresji fenotypowej (1-4). Przerost mięśnia lewej komory najczęściej jest asymetryczny. U około 30% dzieci występuje ograniczony przerost dotyczący tylko jednego segmentu serca. Najczęściej zajęta jest przegroda międzykomorowa i przednio-boczna wolna ściana lewej komory, często występuje przerost koncentryczny i sporadycznie przerost okolicy koniuszka. Kardiomiopatia przerostowa występuje u około 0,2% populacji (1/500 osób) i jest najczęstszą genetycznie uwarunkowaną chorobą układu sercowo-naczyniowego (5). Kardiomiopatia przerostowa jest szczególnie częstą przyczyną nagłej śmierci sercowej (Sudden Cardiac Death – SCD) u dzieci i młodych dorosłych, w tym u wyczynowych sportowców.
Cel pracy
Celem pracy była retrospektywna analiza obrazu klinicznego u dzieci z kardiomiopatią przerostową hospitalizowanych w Klinice Kardiologii Instytutu „Pomnik – Centrum Zdrowia Dziecka”.
Materiał i metody
W okresie od lipca 1991 roku do października 2010 roku w Klinice Kardiologii IP CZD hospitalizowano 85 dzieci, 35 dziewczynek, 50 chłopców, w wieku od 2 miesięcy do 18 lat (średnio 10,7 ± 5,2 lat) z rozpoznaniem kardiomiopatii przerostowej. U wszystkich pacjentów analizowano wywiad rodzinny (występowanie kardiomiopatii przerostowej i nagłych zgonów sercowych u członków rodziny), wywiad dotyczący występowania u pacjenta objawów klinicznych, takich jak omdlenia, zasłabnięcia, ból w klatce piersiowej, duszność oraz wykonano kompleksowe badania kardiologiczne, które obejmowały: badanie przedmiotowe z określeniem klasy wydolności według Nowojorskiego Towarzystwa Kardiologicznego (NYHA), badanie radiologiczne klatki piersiowej, spoczynkowy zapis elekrokardiograficzny (ekg) oraz 24-godzinny metodą Holtera, badanie echokardiograficzne serca (echo), badania metaboliczne, izotopowe (ocena frakcji wyrzutowej komór serca) oraz scyntygrafię perfuzyjną mięśnia sercowego (SPECT) w spoczynku i w wysiłku, próbę wysiłkową z oceną reakcji ciśnienia tętniczego krwi na wysiłek. U każdego pacjenta z kardiomiopatią przerostową zalecono badanie rodziców, rodzeństwa oraz pozostałych członków rodziny w kierunku HCM. W piśmiennictwie przedstawiono opracowany w Klinice Kardiologii IP CZD szczegółowy protokół badań kardiologicznych u pacjentów z kardiomiopatią przerostową (6). Okres obserwacji u pacjentów w analizowanej grupie wynosi od 1 roku do 19,3 lat, (średnio 6,9 lat).
Wyniki
W analizowanej grupie pacjentów rodzinne występowanie kardiomiopatii przerostowej stwierdzono u 41 (51,3%) dzieci. Nagłe zgony sercowe u członków rodziny poniżej 40. roku życia występowały u 24 (28,6%) pacjentów. Kardiomiopatię przerostową w badanej grupie dzieci rozpoznano w wieku od 1 miesiąca życia do 16,7 lat, średnio w wieku 5,9 lat. U 28 (32,9%) dzieci HCM była rozpoznana w wieku niemowlęcym (od 1 do 12 miesiąca życia, średnio w 4,3 miesiącu). Wśród objawów klinicznych pacjenci najczęściej zgłaszali męczliwość, upośledzenie tolerancji wysiłku (57/85 = 67,1% dzieci). W I klasie czynnościowej według NYHA było 39 (46%) pacjentów, w II klasie 38 (45%), w III klasie 7 (8%), natomiast w IV klasie NYHA jeden (1%) pacjent. U 83 (97,6%) dzieci stwierdzano szmer skurczowy nad sercem. Okresowo występujące bóle w klatce piersiowej zgłaszało 26 (30,6%) dzieci. Nawracające omdlenia występowały u 15 (17,6%) pacjentów, natomiast zasłabnięcia (stany przedomdleniowe) u 19 (22,4%) dzieci. Epizody kołatania serca odczuwało 18 (21,2%) pacjentów. W analizowanej grupie dzieci nagłe zatrzymanie krążenia ze skuteczną resuscytacją wystąpiło u 3 (3,5%) pacjentów, natomiast nagły zgon sercowy stwierdzono u 3 (3,5%) dzieci. Objawy kliniczne występujące w analizowanej grupie pacjentów przedstawiono na rycinie 1.
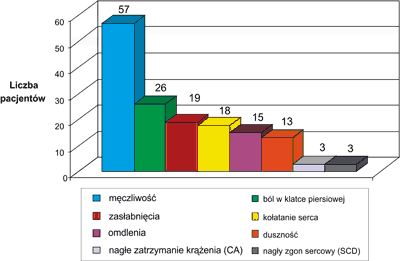
Ryc. 1. Objawy kliniczne występujące u pacjentów z kardiomiopatią przerostową.
W badaniu radiologicznym klatki piersiowej u 42 (49,4%) dzieci stwierdzono powiększoną sylwetkę serca (CTR powyżej 0,50) oraz zastój w krążeniu płucnym u 12 (14,1%) pacjentów.
Nieprawidłowy spoczynkowy zapis ekg stwierdzono u 94,1% pacjentów. U 55 (64,7%) dzieci występowały zmiany odcinka ST i załamka T, u 65 (75,5%) cechy przerostu lewej komory (wskaźnik Sokołowa-Lyona wynosił od 38 mm do 150 mm; norma 37 mm), u 16 (18,8%) dzieci współistniały cechy przerostu mięśnia prawej komory. U 18 (21,2%) pacjentów stwierdzono wydłużenie skorygowanego odstępu QT (QTc powyżej 440), a u 33 (42,3%) dzieci zwiększoną wartość dyspersji QTc powyżej 50 ms.
W 24-godzinnym zapisie ekg metodą Holtera u 11 (12,9%) dzieci stwierdzano epizody nieutrwalonego częstoskurczu komorowego (nsVT), natomiast u żadengo pacjenta nie występował utrwalony częstoskurcz komorowy. U 39 (45,9%) dzieci stwierdzono komorowe zaburzenia rytmu serca pod postacią dodatkowych pobudzeń komorowych, w tym u 4 dzieci powyżej 1000/dobę. Napadowy częstoskurcz nadkomorowy zarejestrowano u 4 (4,7%) badanych. U 1 dziecka (1,2%) ze znacznie powiększonym wymiarem lewego przedsionka rozpoznano migotanie przedsionków.
W badaniu echo serca u 53 (62,4%) pacjentów stwierdzono przerost asymetryczny mięśnia lewej komory, u 32 (37,6%) przerost koncentryczny mięśnia lewej komory, natomiast u 2 (2,4%) przerost części koniuszkowej lewej komory. Grubość przegrody międzykomorowej wynosiła średnio 15,6 mm (od 4,3 mm do 44 mm), co w odniesieniu do powierzchni ciała (BSA) dziecka stanowiło 248,4% średniej normy dla BSA (od 105 do 657% średniej normy dla BSA). Istotny przerost mięśnia lewej komory (powyżej 190% średniej normy w stosunku do BSA) stwierdzono u 59 (69,4%) pacjentów, zaś masywny przerost powyżej 30 mm u 8 (8,5%) pacjentów. Grubość tylnej ściany lewej komory wynosiła średnio 8,9 mm (wahała się od 4,0 mm do 20,7 mm), co w odniesieniu do powierzchni ciała stanowiło średnio 142,2% normy. Przerost mięśnia prawej komory współistniał u 17 (20%) pacjentów. Postać zawężającą kardiomiopatii przerostowej (wielkość gradientu w drodze odpływu lewej komory powyżej 30 mmHg w spoczynku) stwierdzono u 20 (23,5%) pacjentów.
W badaniu izotopowym serca u 6/67 (9%) pacjentów stwierdzono obniżoną frakcję wyrzutową lewej komory serca, natomiast u 6 (9%) obniżoną również frakcję wyrzutową prawej komory.
Badanie perfuzji mięśnia sercowego metodą SPECT wykonano w spoczynku u 57 (67,1%) pacjentów, niedokrwienie mięśnia sercowego stwierdzono u 18 (18/57 = 31,6%) dzieci. Badanie perfuzji mięśnia sercowego metodą SPECT w wysiłku wykonano u 49 pacjentów (57,6%), niedokrwienie mięśnia sercowego stwierdzono u 34 (34/49 = 69,4%) dzieci. U 16 (28%) pacjentów nie występowały cechy niedokrwienia mięśnia sercowego.
Próbę wysiłkową metodą Bruce’a na bieżni ruchomej wykonano u 50 pacjentów. U 17 (34%) spośród nich stwierdzono nieprawidłową reakcję ciśnienia tętniczego na wysiłek (u 10 dzieci spadek ciśnienia tętniczego na szczycie wysiłku, natomiast u 7 pacjentów brak wzrostu ciśnienia w czasie wysiłku).
Nieprawidłowe wartości badań metabolicznych stwierdzono u 50/72 (69,4%) pacjentów: u 30% dzieci stwierdzano obniżony poziom karnityny wolnej i całkowitej w moczu oraz w surowicy, u 8 pacjentów nieprawidłowy test metaboliczny, nieprawidłowy profil kwasów organicznych w moczu u 5 dzieci, izoformy transferyny u 1 pacjenta, a u 6 dzieci stwierdzono podwyższony poziom kwasu mlekowego we krwi. U wszystkich dzieci z HCM przeprowadzono konsultacje metaboliczne w Poradni Metabolicznej IP CZD w celu dalszej diagnostyki i leczenia.
Leczenie farmakologiczne włączono u 83 (97,6%) dzieci. Spośród nich 70 (82,4%) pacjentów otrzymuje beta-blokery w średniej dawce 0,9 mg/kg mc/dobę, natomiast 12 (14,1%) dzieci blokery kanału wapniowego (średnia dawka 2,1 mg/kg mc/dobę). Pacjenci z HCM oraz z objawami niewydolności serca wymagali leczenia objawowego niewydolności serca. Leczenie antyarytmiczne stosowano u 4 dzieci (Amiodaron u 1 dziecka, Sotalol u 3 pacjentów).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Maron BJ: Hypertrophic cardiomyopathy in childhood. Paediatr Clin North Am 2004; 51(5): 1305-134
2. Ziółkowska L, Kawalec W, Turska-Kmieć A: Kardiomiopatia przerostowa – odrębności wieku dziecięcego. Kardiol Dypl 2006; 4: 44-52.
3. Nugent A, Daubeney P, Chondros P et al.: Clinical features and outcomes of childhood hypertrophic cardiomyopathy: results from a national population-based study. Circulation 2005; 112: 1332-1338.
4. Ziółkowska L, Kawalec W, Pręgowska K et al.: Hypertrophic cardiomyopathy in children - clinical profile in different type of left ventricular hypertrophy. Kardiol Pol 2007; 65 (8) supl. 3: 110-111.
5. Maron B: Hypertrophic cardiomyopathy: a systematic review. JAMA 2002; 287: 1308-1320.
6. Ziółkowska L, Kawalec W, Turska-Kmieć A et al.: Postępy w diagnostyce i leczeniu kardiomiopatii przerostowej u dzieci. Standardy Medyczne 2006; 3, 2: 147-151.
7. Östman-Smith I, Wettrell G, Keeton B et al.: Age-and gender-specific mortality rates in childhood hypertrophic cardiomyopathy. Eur Heart J 2008; 29: 1160-1167.
8. Yetman A, Hamilton R, Benson L, McCrindle B: Long-term outcome and prognostic determinants in children with hypertrophic cardiomyopathy. J Am Coll Cardiol 1998; 32: 1943-1950.
9. Wilkinson J, Sleeper L, Alvarez J et al.: The Pediatric Cardiomyopathy Registry: 1995-2007. Prog Pediatr Cardiol 2008; 25(1): 31-36.
10. Colan S, Lipshultz S, Lowe A et al.: Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: findings from the pediatric cardiomyopathy registry. Circulation 2007; 115: 773-781.
11. Decker J, Rossano J, O’Brian Smith E et al.: Risk factors and mode of death in isolated hypertrophic cardiomyopathy in children. J Am Coll Cardiol 2009; 54: 250-254.
12. Ostman-Smith I: Hypertrophic cardiomyopathy in childhood and adolescence – strategies to prevent sudden death. Fundamental & Clinical Pharmacology 2010; 24: 637-652.
13. Corrado D, Basso C, Schiavon M et al.: Screening for hypertrophic cardiomyopathy in young athletes. N Engl J Med 1998; 339: 364-369.
14. Montgomery J, Gohman T, Harris K et al.: Electrocardiogram in hypertrophic cardiomyopathy revisited: does ECG pattern predict phenotypic expression and left ventricular hypertrophy or sudden death? J Am Coll Cardiol 2002; 39 (Suppl A): 161A.
15. Esteban M, Kaski J: Hypertrophic cardiomyopathy in children. Pediatrics And Child Health 2007; 17:19-24.
16. Östman-Smith I, Wettrell G, Keeton B et al.: Echocardiographic and electrocardiographic identification of those children with hypertrophic cardiomyopathy who should be considered at high-risk of dying suddenly. Cardiol Young 2005; 15: 632-642.
17. McKenna W: The natural history of hypertrophic cardiomyopathy. Cardiovasc Clin 1988; 19: 135-148.
18. Zipes DP, Camm AJ, Borggrefe M et al.: ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Europace 2006; 8: 746-837.
19. Dimitrow P, Chojnowska L, Rudziński T et al.: Sudden death in hypertrophic cardiomyopathy: old risk factors re-assessed in a new model of maximalized follow-up. Eur Heart J 2010; ehq308v1.
20. Maron B: Risk Stratification and Role of Implantable Defibrillators for Prevention of Sudden Death in Patients with Hypertrophic Cardiomyopathy. Circ J 2010; 74: 2271-2282.
21. McKenna W, Deanfield J: Hypertrophic cardiomyopathy: an important cause of sudden death. Arch Dis Child 1984; 59: 971-975.
22. Ziółkowska L, Kawalec W, Turska-Kmieć A et al.: Czynniki ryzyka nagłego zgonu sercowego u dzieci z kardiomiopatią przerostową. Standardy Med 2009; 6, 5: 823-828.
23. Ziółkowska L, Kawalec W, Turska-Kmieć A et al.: Identification of high-risk patients among children with hypertrophic cardiomyopathy. Cardiol Young 2008; 18: 1-19.
24. Colan S: Hypertrophic cardiomyopathy in childhood. Heart Fail Clin 2010; 6(4): 433-44.