© Borgis - Postępy Nauk Medycznych 9/2012, s. 699-704
*Krzysztof Jamroziak1, Olga Grzybowska-Izydorczyk2, Anna Szmigielska-Kapłon1, Dorota Jesionek-Kupnicka3, Ewa Wawrzyniak1, Barbara Pieńkowska-Grela4, Tadeusz Robak1
Nowotwory z blastycznych plazmacytoidalnych komórek dendrytycznych – opis dwóch przypadków
Blastic plasmacytoid dendritic cell neoplasms – a report of two cases
1Department of Hematology, Medical University of Łódź
Head of Department: prof. Tadeusz Robak, MD, PhD
2Department of Experimental Hematology, Medical University of Łódź
Head of Department: prof. Piotr Smolewski, MD, PhD
3Department of Pathology, Medical University of Łódź
Head of Department: prof. Radzisław Kordek, MD, PhD
4Department of Cytogenetic, The Maria Skłodowska-Curie Memorial Cancer Center and Institute, Warsaw
Head of Department: Barbara Pieńkowska-Grela, MD, PhD
Streszczenie
Artykuł zawiera opis dwóch przypadków chorych z rozpoznaniem bardzo rzadko występujących nowotworów z blastycznych plazmacytoidalnych komórek dendrytycznych (blastic plasmacytoid dendritic cell neoplasms – BPDCN). BPDCN charakteryzują się koekspresją antygenów CD4 i CD56 przy braku ekspresji innych mieloidalnych lub limfoidalnych markerów liniowych, pierwotnym zajęciem skóry i wtórnym szpiku kostnego oraz agresywnym przebiegiem klinicznym. Rokowanie jest złe, szczególnie w przypadku leczenia konwencjonalną chemioterapią. Zgodnie z opublikowanymi danymi jedyną opcją terapeutyczną, która wydaje się poprawiać rokowanie w BPDCN jest intensywna chemioterapia skojarzona z allogenicznym przeszczepieniem macierzystych komórek hematopoetycznych.
Summary
In this paper we report two cases of very rare blastic plasmacytoid dendritic cell neoplasms (BPDCN). BPDCN are characterized by co-expression of CD4 and CD56 antigens in the absence of any specific myeloid or lymphoid lineage or markers, primary skin infiltrations followed by bone marrow involvement, and aggressive clinical course. The prognosis is poor, especially when treatment is based on conventional chemotherapy. According to published evidence the only treatment with some potential to improve BPDCN outcome is intensive chemotherapy consolidated by allogeneic hematopoietic stem cell transplantation.

Introduction
A category of rare and aggressive haematologic tumors co-expressing CD4 and CD56 antigens in the absence of any specific myeloid, B-, T-lymphoid or natural killer (NK) lineage markers is classified as blastic plasmacytoid dendritic cell neoplasms (BPDCN) according to the most recent 2008 WHO classification of tumours of haematopoietic and lymphoid tissues (1). This is in line with the recent knowledge that BPDCN are derived from the precursors of plasmacytoid dentritic cells (2-4). However, in the past a number of different terms were used for BPDCN due to their uncommon immunophenotypic, histopathological and clinical features, including blastic NK leukemia/lymphoma, histiocytic lymphoma or histiocytic associated hematologic malignancy, cutaneous a granular CD4+ CD56+ hematodermic neoplasm and NK-cell lymphoma or myelo-monocytic precursor – related lymphoma (5-12).
BPDCN are diagnosed about three times more often among men than women, mainly in the elderly (median age at diagnosis at 7th decade of live) (5, 10). Till present the etiology of BPDCN has not been elucidated. Epstein Barr virus and HIV had been previously supposed to play a role in BPDCN pathogenesis, but later reports did not confirm their correlation to the disease development (5-12). Clinically, the typical initial presentation of BPDCN is presence of asymptomatic cutaneous lesions that are subsequently followed by bone marrow involvement and leukemic dissemination (5, 10). The disease is characterized by highly aggressive clinical course. Although BPDCN respond to different types of chemotherapy, the responses are usually of short duration, and disease is most often fatal within a few years from diagnosis (5, 10).
Despite rare incidence of BPDCN, numerous variations from typical clinicopathological picture of BPDCN have been reported, such as different extracutaneous manifestation, lack of skin involvement, immunophenotypic variants or other, making a diagnosis even more complicated (13-17). In this paper we report two cases of BPDCN diagnosed in our center with objective to increase clinical awareness for these rare and aggressive malignancies that seem to be underdiagnosed in Poland.
Case 1
63-year old man was referred to the Department of Chemotherapy of Medical University of Łódź in April 2008 for evaluation of potentially malignant skin lesions. The history of B symptoms was negative. On examination, the skin lesions were found to be predominantly purpuric nodules, in association with bruise-like areas, and were distributed over the left forearm. The majority of the nodules ranged from 1 to 2 cm in diameter. The nodules were not painful on palpation and the temperature of the adjacent skin was normal. On the physical examination lymphadenopathy, hepatomegaly or splenomagaly were excluded. The computed tomography scan of the chest and abdomen revealed no enlarged lymph nodes in the mediastinum or retroperitoneum. The complete blood count (CBC) was normal.
On the basis of histopathological and immunohistochemical examination of the full-layer skin biopsy the diagnosis of BPDCN was established (fig. 1A-D). The skin was shown to be involved by dense, diffuse infiltration of blastic cells with irregular nuclei and one to a few nucleoli (fig. 1D).
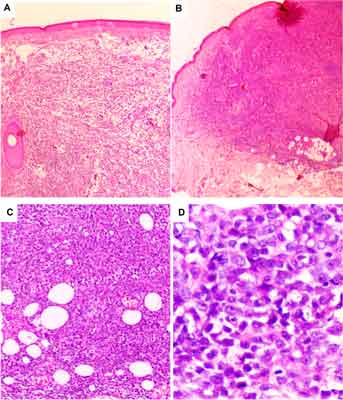
Fig. 1. The histopathological features of blastic plasmacytoid dendritic cell neoplasm infiltrating the skin in the case 1 (A, C) and case 2 (B, D). The infiltrates are dense and diffuse, save the epidermis (A) but deeply invade the adipose tissue (B and C). The blastic cells are medium-sized with variable shape of nuclei and various number of nucleoli; the mitotic figures are seen (D).
The epidermis was intact, while the infiltration was extended to subcutaneous fat (fig. 1 A, B, C). The variable number of mitoses was seen (fig. 1D). Immunohistochemical profile was determined as positive for LCA, CD4, CD43 and CD56, and negative for such lineage markers as CD3, CD8, CD20, CD30, myeloperoxidase (MPO) and terminal deoxynucleotidyltransferase (TdT) andgranzyme B. For differential diagnosis with other small blue round cell tumors staining with other markers were also performed, namely CD99 (-), CKMNF (-), CK20 (-), HMB45 (-), NSE-, TTF1 (-), CKAE1/AE3 (-), Melan A (-), Vimentin (+), and synaptophysin (-). Proliferative index of Ki -67 (MIB-1) reached 80%. Additionally, bone marrow aspiration was performed to exclude bone marrow involvement by BPDCN and shown normal marrow cytology. The treatment was based on standard chemotherapy protocol CHOP (cyclophosphamide, doxorubicine, vincristine and prednisone) protocol. The patient received seven CHOP cycles at 4-week intervals and achieved complete resolution of skin infiltrations.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Swerdlow SH, Campo E, Harris NL et al.: WHO classification of tumours of haematopoietic and lymphoid tissues, Fourth Edition. 2008.
2. Petrella T, Comeau MR, Maynadié M et al.: ‘Agranular CD4+ CD56+ hematodermic neoplasm’ (blastic NK-cell lymphoma) originates from a population of CD56+ precursor cells related to plasmacytoid monocytes. Am J Surg Pathol 2002; 26: 852-862.
3. Jaye DL, Geigerman CM, Herling M et al.: Jones D. Expression of the plasmacytoid dendritic cell marker BDCA-2 supports a spectrum of maturation among CD4+ CD56+ hematodermic neoplasms. Mod Pathol 2006; 19: 1555-1562.
4. Urosevic M, Conrad C, Kamarashev J et al.: CD4+CD56+ hematodermic neoplasms bear a plasmacytoid dendritic cell phenotype. Hum Pathol 2005; 36: 1020-1024.
5. Jacob MC, Chaperot L, Mossuz P et al.: CD4+ CD56+ lineage negative malignancies: a new entity developed from malignant early plasmacytoid dendritic cells. Haematologica 2003; 88 (8): 941-955.
6. Herling M, Jones D: CD4+/CD56+ hematodermic tumor: the features of an evolving entity and its relationship to dendritic cells. Am J Clin Pathol 2007; 127: 687-700.
7. Kameoka J, Ichinohasama R, Tanaka M et al.: A cutaneous agranular CD2- CD4+ CD56+ „lymphoma”: report of two cases and review of the literature. Am J Clin Pathol 1998; 110: 478-488.
8. Ascani S, Massone C, Ferrara G et al.: CD4-negative variant of CD4+/CD56+ hematodermic neoplasm: description of three cases. J Cutan Pathol 2008; 35: 911-915.
9. Weaver J, Hsi ED: CD4+/CD56+ hematodermic neoplasm (blastic NK-cell lymphoma). J Cutan Pathol 2008; 35: 975-977.
10. Cota C, Vale E, Viana I et al.: Cutaneous manifestations of blasticplasmacytoid dendritic cell neoplasm-morphologic and phenotypic variability in a series of 33 patients. Am J Surg Pathol 2010; 34: 75-87.
11. Pilichowska ME, Fleming MD, Pinkus JL, Pinkus GS: CD4+/CD56+ hematodermic neoplasm („blastic natural killer cell lymphoma”): neoplastic cells express the immature dendritic cell marker BDCA-2 and produce interferon. Am J Clin Pathol 2007; 128: 445-453.
12. Piña-Oviedo S, Herrera-Medina H, Coronado H et al.: CD4+/CD56+ hematodermic neoplasm: presentation of 2 cases and review of the concept of an uncommon tumor originated in plasmacytoid dendritic cells expressing CD123 (IL-3 receptor alpha). Appl Immunohistochem Mol Morphol 2007; 15: 481-486.
13. Shiman M, Marchione R, Ricotti C et al.: CD4+/CD56+ Hematodermic neoplasm (plasmacytoid dendritic cell tumor). Dermatol Online J 2008; 15: 5.
14. Giagounidis AA, Heinsch M, Haase S, Aul C: Early plasmacytoid dendritic cell leukemia/lymphoma coexpressing myeloid antigenes. Ann Hematol 2004; 83: 716-721.
15. Li Y, Li Z, Lin HL et al.: Primary cutaneous blasticplasmacytoid dendritic cell neoplasm without extracutaneous manifestation: case report and review of the literature. Pathol Res Pract 2011; 207: 55-59.
16. Rauh MJ, Rahman F, Good D et al.: Blasticplasmacytoid dendritic cell neoplasm with leukemic presentation, lacking cutaneous involvement: Case series and literature review. Leuk Res 2012; 36: 81-86.
17. Stetsenko GY, McFarlane R, Kalus A et al.: CD4+/CD56+ hematodermic neoplasm: report of a rare variant with a T-cell receptor gene rearrangement. J Cutan Pathol 2008; 35: 579-584.
18. Patel JL, Shetty S, Salama ME: An unusual case of cutaneous blasticplasmacytoid dendritic cell neoplasm with concomitant B-cell lymphoproliferative disorder. Am J Dermatopathol 2011; 33: 31-36.
19. Shaffer LG, Slovak ML, Campbell LJ: ISCN 2009: An International System for Human Cytogenetic Nomenclature (2009). Basel: S. Karger; 2009.
20. Holowiecki J, Grosicki S, Robak T et al.: Polish Adult Leukemia Group (PALG). Addition of cladribine to daunorubicin and cytarabine increases complete remission rate after a single course of induction treatment in acute myeloid leukemia. Multicenter, phase III study. Leukemia 2004; 18: 989-997.
21. Robak T, Blonski JZ, Gora-Tybor J et al.: Polish Leukemia Group (PALG CLL2). Cladribine alone and in combination with cyclophosphamide or cyclophosphamide plus mitoxantrone in the treatment of progressive chronic lymphocytic leukemia: report of a prospective, multicenter, randomized trial of the Polish Adult Leukemia Group (PALG CLL2). Blood 2006; 108: 473-479.