© Borgis - Medycyna Rodzinna 3/2012, s. 51-55
*Paweł Kowalczyk1, Dorota Dziuban2, Katarzyna Hulka3, Maciej Filocha1
Zastosowanie różnorodnych metod bioinformatycznych w badaniach biologicznych i medycznych
Using a varienty of bioinformatic methods in biological and medical research
1 Interdyscyplinarne Centrum Modelowania Matematycznego i Komputerowego Uniwersytetu Warszawskiego
Dyrektor Centrum: prof. dr hab. Marek Niezgódka
2Instytut Biochemii i Biofizyki Polskiej Akademii Nauk, Zakład Biologii Molekularnej
Dyrektor Instytutu: prof. dr hab. Piotr Zielenkiewicz
Kierownik Zakładu: prof. dr hab. Barbara Tudek
3Wydział Biologii Uniwersytetu Warszawskiego
Dziekan Wydziału: prof. dr hab. Joanna Pijanowska
Summary
Examination of potential mutagenic and carcinogenic properties of new products entering the market or to the environment that may induce human cancers has been made compulsory for the various sectors of the economy in the various fields as medicine and molecular biology. The number of new chemical carcinogenic compounds present in the human environment with the ability of mutations in critical genes (oncogenes or tumor suppressor genes) requires the formation of ever new methods, techniques and screening tests used in the micro-organisms (bacteria, yeast, viruses, cell culture) or higher organisms (fruit fly). Studies in animals (rodents) are carried out only in the final testing phase of the compound. A very large amount of information required for pre-selection of active compounds) was frequently replaced in silico studies. Therefore, with the help of bioinformatics has developed a number of computational programs (information and math), explaining in a simple way to very complex biological systems and medical devices within the entire genome, because they are much more sensitive and they can study the changes in organisms exposed to low doses of mutagens or diversity within the population. The ability to process a gigantic amount of information obtained during the study of genomes and proteomes of living organisms is essential not only in scientific work, but also for practical applications as the design of therapeutic substances, crystallochemistry drugs, magnetic resonance spectroscopy in biological research, quantum chemistry and molecular modeling, instrumental analysis used in organic synthesis of classical and molecular biology.

Modelowanie matematyczne w służbie nauk biologicznych i medycznych
Obecnie modelowanie zachowania się układów biologicznych odgrywa ogromne znaczenie w biologii molekularnej, syntetycznej biochemii i medycynie. Wiele metod matematycznych znalazło zastosowanie do symulowania zachowania działania układów badanych przez te dyscypliny, jak: przemiany metaboliczne zachodzące w komórce, wpływ kancerogenów chemicznych na stabilność helisy DNA, projektowanie nowych leków związanych z chorobami autoregeneracyjnymi. Dostępnych jest wiele darmowych narzędzi programistycznych umożliwiających prowadzenie takich symulacji. Niestety wraz ze wzrostem złożoności układu drastycznie wzrasta złożoność obliczeniowa symulacji oraz stopień nieprzewidywalności zachowania się układu. Dodatkowym utrudnieniem w tworzeniu poprawnie działających modeli jest duża liczba czynników zewnętrznych, które zakłócają jego działanie a często nie są należycie uwzględniane (bądź nie są znane) podczas konstruowania takich modeli. Tworzenie matematycznych modeli opisujących projektowany układ pozwala na przewidzenie niektórych jego cech, co może posłużyć do jego udoskonalenia w celu wyeliminowania wad lub wprowadzenia pewnych usprawnień. Obecnie zostały rozwinięte wielkoskalowe modele sieci genetycznych tworzone z myślą o zastosowaniach w sekwencjonowaniu metagenomów ssaczych i bakteryjnych. Pozwalają one na odtwarzanie podstawowych procesów biologicznych, takich jak transkrypcja, translacja oraz wpływ czynników transkrypcyjnych oraz sygnałów środowiskowych na działanie całych sieci genetycznych. Do prowadzenia takich symulacji można wykorzystać różnorodne oprogramowanie komputerowe. Do często stosowanych programów należą: matlab – nazwa programu pochodzi od angielskich słów MATrix LABoratory, gdyż początkowo program ten był przeznaczony do numerycznych obliczeń macierzowych. Cechuje go duża liczba funkcji bibliotecznych oraz duże możliwości rozbudowy przez użytkownika za pomocą pisania własnych funkcji. Posiada on swój język programowania, co umożliwia pisanie w pełni funkcjonalnych programów działających w środowisku Matlaba. W zakresie grafiki Matlab umożliwia rysowanie dwu- i trójwymiarowych wykresów funkcji oraz wizualizację wyników obliczeń w postaci rysunków statycznych i animacji. Możliwe jest pobieranie danych pomiarowych z urządzenia zewnętrznego przez porty w celu ich obróbki. Wszystko to powoduje, że program ten znajduje bardzo szerokie zastosowanie w medycynie i biologii molekularnej. Istnieją także alternatywne odpowiedniki tego programu, takie jak Scilab, Promot, CellDesigner czy Octave – służą do prostych obliczeń przez macierze, algebrę liniową, przetwarzanie sygnałów, przy wykorzystaniu środowiska graficznego są w stanie rysować grafy i wykresy dwu- i trzywymiarowe, a nawet tworzyć animacje. Do tego typu obliczeń statystycznych służy pakiet STATISTICA, który zawiera bardzo dużą liczbę metod obliczeniowych do analizowania danych zebranych w eksperymentach, jak i do dopasowywania planów do ciągłych i skategoryzowanych zmiennych z wykorzystaniem kompletnej analizy wariancji typu (ANOVA). Użytkownik sprawuje pełną kontrolę nad tym, które efekty i interakcje będą włączone do analizowanego modelu; może przeglądać korelacje pomiędzy kolumnami macierzy planu (X) i zapoznać się z odwrotnością macierzy iloczynowej X’X (tzn. udostępniane są macierze korelacji i kowariancji estymatorów parametrów). W programie obliczane są estymatory parametrów ANOVA, ich odchylenia standardowe oraz przedziały ufności, a także współczynniki regresji (odchylenia standardowe, przedziały ufności) zarówno dla unormowanych (przedział [-1, +1]), jak i rzeczywistych wielkości wejściowych. Na podstawie wspomnianych estymatorów program umożliwia obliczenie wartości aproksymowanych (oraz odchyleń standardowych i przedziałów ufności) dla zadanych przez użytkownika wartości wielkości wejściowych. Liczne opcje umożliwiają graficzne przedstawienie uzyskanych wyników: wykresy Pareto efektów, wykresy prawdopodobieństwa normalnego i normalnego połówkowego efektów, wykresy kwadratowe i sześcienne, wykresy wartości średnich i interakcji (wraz z przedziałami ufności dla średnich krańcowych), wykresy przestrzenne i warstwicowe powierzchni odpowiedzi (ryc. 1).
Ryc. 1. Przykładowy rzut ekranu.
Program Multi Gauge wersja 3.0 wyprodukowany przez firmę FUJIFILM, służy do analizy wyników graficznych w postaci prążków z żeli agarozowych i akrylamidowych wymagających obróbki ilościowej. Do głównych funkcji programu należą: korekta obrazu (Display) umożliwiająca regulację kontrastu, obróbka obrazu (Process): obrót względem osi, redukcja szumu, umieszczenie adnotacji (Annotation): komentarzy i strzałek, pomiar (Measure): obrazu żeli, płytek, kalibrację (Calibration): nanoszenie linii kalibracyjnych dla stężenia i masy molekularnej, analizę wyników (Analysis). Przykładową analizę obrazu żelu przedstawiono na rycinie 2.
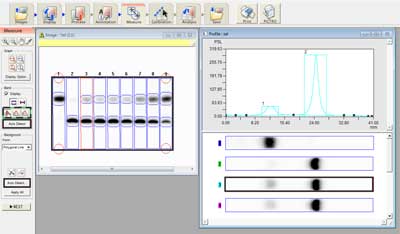
Ryc. 2. Przykładowa analiza obrazu żelu. Doświadczenie miało na celu pomiar aktywności enzymu, którą odzwierciedla procentowy udział prążka o mniejszej masie. W celu uzyskania wyraźnego sygnału i zmniejszenia tła, dopasowano kontrast i jasność obrazu. Aby otrzymać profil każdej ze studzienek przeprowadzono autodetekcję prążków i tła (linia pogrubiona), w razie potrzeby naniesiono własne poprawki (linia przerywana).
Alternatywne procedury analizowania danych doświadczalnych
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Tornaletti S, Pfeifer GP: Slow repair of pyrimidine dimers at p53 mutation hotspots in skin cancer. Science 11 Mar 1994; 263 (5152): 1436-1438. 2. Pfeifer GP, Tornaletti S: Footprinting with UV irradiation and LMPCR. Methods Feb 1997; 11 (2): 189-196. 3. Pfeifer GP, Denissenko MF, Tang MS: PCR-based approaches to adduct analysis. Toxicol Lett 28 Dec 1998; 102-103: 447-451. 4. Pfeifer GP, Dammann R: Measuring the formation and repair of UV photoproducts by ligationmediated PCR. Methods Mol Biol 1999; 113: 213-226. 5. Smith LE, Denissenko MF, Bennett WP et al.: Targeting of lung cancer mutational hotspots by polycyclic aromatic hydrocarbons. J Natl Cancer Inst 2000; 92: 803-811. 6. Lau AY, Scharer OD, Samson L et al.: Crystal structure of a human alkylbase-DNA repair enzyme complexed to DNA: Mechanisms for Nucleotide flipping and base excision. Cell 16 Oct 1998; 95: 249-258. 7. Stivers JT, Pankiewicz KW, Watanabe KA: Kinetic mechanism of damage site recognition and uracil flipping by Escherichia coli uracil DNA glycosylase. Biochemistry 1999; 38: 952-963. 8. Hollis T, Ichikawa Y, Ellenberger T: DNA bending and a flip-out mechanism for base excision by the helix-hairpin-helix DNA glycosylase, Escherichia coli AlkA. EMBO J 2000; 19: 758-766. 9. Dodson ML, Michaels ML, Lloyd RS: Unified catalytic mechanism for DNA glycosylases. J Biol Chem 1004; 269: 32709-32712. 10. Krokan HE, Standal R, Slupphaug G: DNA glycosylases in the base excision repair of DNA. Biochem J 1997; 325: 1-16. 11. Mitra S: DNA glycosylases: specificity and mechanisms. Prog Nucleic Acid Res Mol Biol 2001; 68: 189-192. 12. Scharer OD, Jiricny J: Recent progress in the biology, chemistry and structural biology of DNA glycosylases. Bioessays 2001; 23: 270-281. 13. Matsumoto Y, Kim K, Bogenhagen DF: Proliferating cell nuclear antigen-dependent abasic site repair in Xenopus laevis oocytes: an alternative pathway of base excision DNA repair. Mol Cell Biol 1994; 14: 6187-6197. 14. Frossina G, Fortini P, Rossi O et al.: Two pathways for base excision repair in mammalian cells. J Biol Chem 1996; 271: 9573-9578. 15. Mohn GR: Bacterial systems for carcinogenicity testing. Mutation Research 1981; 87: 191-210.
