© Borgis - Nowa Pediatria 4/2012, s. 81-83
*Katarzyna Wójcicka, Zdzisław Domagała
Kliniczne rozpoznanie zespołu Townesa-Brocksa u niemowlęcia – opis przypadku
Clinical diagnosis of Townes-Brocks syndrome in an infant – a case report
II Oddział Chorób Dziecięcych Wojewódzkiego Specjalistycznego Szpitala Dziecięcego im. Władysława Buszkowskiego w Kielcach
Kierownik Oddziału: dr n. med. Zdzisław Domagała
Summary
Townes-Brocks syndrome (TBS) is a rare multiple malformation syndrome with clinical variability. The diagnosis is based on the presence of malformations of the ears, limbs, kidneys and urinary tract and anus. Disease results from mutation in SALL1 gene, although its absence does not exclude the diagnosis. The article reports a 8-month-old infant with dysplastic ears, bilateral hearing loss, triphalangeal thumbs, overlapping fingers and toes and vesicoureteral reflux into the solitary functioning left kidney. Congenital anomalies seen in the members of the family confirm phenotypic variability of this condition.

WSTĘP
Nazwa zespołu wywodzi się od nazwisk autorów, którzy w 1972 roku po raz pierwszy opisali wady wrodzone u ojca i jego pięciorga dzieci (1). Charakteryzuje się on triadą objawów: niedrożnością odbytu, dysplazją uszu i malformacjami kciuka, lecz opisywano także szerokie spektrum dodatkowych wad (wady nerek, dłoni i stóp, serca, oczu, upośledzenie słuchu) (2,3). Jedynym udokumentowanym genem związanym z zespołem jest SALL1 – ludzki homolog genu spalt (sal) Drosophili melanogaster, zlokalizowany na chromosomie 16q12.1 (3-5). Aktualnie znanych jest ponad 60 jego mutacji (6). Choroba dziedziczy się autosomalnie dominująco (2-6).
OPIS PRZYPADKU
Dziewczynka aktualnie 8-miesięczna z ciąży drugiej, urodzona cięciem cesarskim (stan po cięciu cesarskim) w 39 hbd, z ciężarem ciała 3000 g, Apgar 8/9 pkt (w badaniu fizykalnym u noworodka stwierdzono trójpaliczkowe kciuki i dysmorficzne uszy). W wieku 2 miesięcy została skierowana do Wojewódzkiego Specjalistycznego Szpitala Dziecięcego w Kielcach celem diagnostyki radiologicznej układu moczowego z powodu agenezji nerki prawej w badaniu ultrasonograficznym. W wywiadzie rodzinnym: ojciec zdrowy, matka po korekcji chirurgicznej lewej dysplastycznej małżowiny usznej, rodzina matki (wujek – jedyny brat matki, babcia dziecka i jej siostra z dwiema córkami) z wadami małżowin usznych. U 6-letniego brata pacjentki, będącego pod opieką Poradni Genetycznej (kariotyp prawidłowy, badania mutacji w genie SALL1 nie wykonywano), obecne są liczne nieprawidłowości rozwojowe (przesunięcie odbytu ku przodowi, obustronny niedosłuch z niedorozwojem małżowiny usznej prawej, dysplazja wielotorbielowata nerki lewej oraz wady kończyn górnych – aktualnie po kilkuetapowej korekcji). Przy przyjęciu dziewczynka w stanie ogólnym dobrym, badaniem fizykalnym z odchyleń od normy stwierdzano wady wrodzone obu uszu (małe, nisko osadzone, z towarzyszącym niewielkim zagięciem górnej części obrąbka) (ryc. 1), kończyn górnych (trójpaliczkowe kciuki, zachodzenie drugiego na trzeci palec dłoni lewej) (ryc. 2) i dolnych (zachodzenie drugiego na trzeci palec stopy prawej) (ryc. 3). Rozwój psychoruchowy, masa ciała i wzrost odpowiadały normom dla wieku. Wykonane ponownie usg potwierdziło agenezję nerki prawej, a w nerce lewej (o wymiarach 56 x 25 mm) widoczne było światło miedniczki (V-0,2 ml). Cystografia mikcyjna wykazała lewostronny odpływ pęcherzowo-moczowodowy bierny II/III oraz czynny IV stopnia (ryc. 4). W renoscyntygrafii uwidoczniono jedyną nerkę lewą o typowym położeniu i prawidłowej czynności, ale z niewielkimi ubytkami kory w tomografii SPECT, mogącymi odpowiadać zmianom pozapalnym (bez zakażeń układu moczowego w wywiadzie). Wykładniki czynności nerek oraz ciśnienie tętnicze mieściły się w granicach normy. Aktualnie dziecko jest zakwalifikowane do zabiegu ostrzyknięcia ujścia pęcherzowego lewego moczowodu. Pozostaje pod stałą kontrolą Poradni Nefrologicznej i Urologicznej, a z powodu obustronnego umiarkowanego niedosłuchu odbiorczego w Poradni Audiologicznej. W badaniu okulistycznym i kardiologicznym nie stwierdzono odchyleń od normy. Oznaczony w Poradni Genetycznej kariotyp jest prawidłowy, żeński (46, XX), natomiast dotychczas nie zgłosiła się na zaplanowane badanie mutacji SALL1.
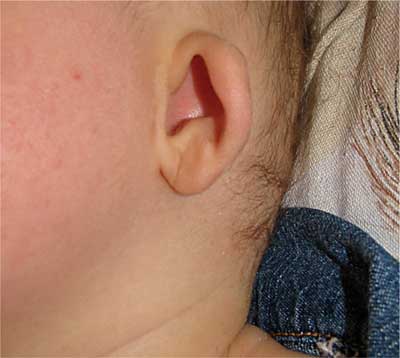
Ryc. 1. Dysplastyczne uszy u pacjentki z TBS.

Ryc. 2. Dłonie: obustronne trójpaliczkowe kciuki i zachodzenie drugiego na trzeci palec ręki lewej.
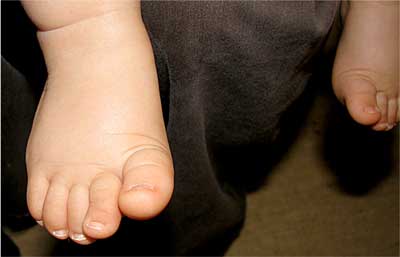
Ryc. 3. Stopa prawa – zachodzenie drugiego na trzeci palec.
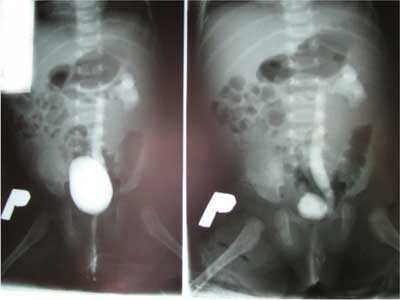
Ryc. 4. Cystografia mikcyjna: wsteczny odpływ pęcherzowo-moczowodowy do jedynej nerki lewej.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Townes PL, Brocks ER: Hereditary syndrome of imperforate anus with hand, foot, and ear anomalies. J Pediatr 1972; 81: 321-326. 2. O’Callaghan M, Young ID: The Townes-Brocks syndrome. J Med Genet 1990; 27(7): 457-461. 3. Powell CM, Michaelis RC: Townes-Brocks syndrome. J Med Genet 1999; 36(2): 89-93. 4. Botzenhart EM, Green A, Ilyina H et al.: SALL1 mutation analysis in Townes-Brocks syndrome: twelve novel mutations and expansion of the phenotype. Hum Mutat 2005; 26(3): 282. 5. Kohlhase J: SALL1 mutations in Townes-Brocks syndrome and related disorders. Hum Mutat 2000; 16(6): 460-466. 6. Miller EM, Hopkin R, Bao L, Ware SM: Implications for genotype-phenotype predictions in Townes-Brocks syndrome: case report of a novel SALL1 deletion and review of the literature. Am J Med Genet A 2012; 158A(3): 533-540. 7. Kowalewska-Pietrzak M, Młynarski W: Dziecko z wrodzonymi mnogimi wadami układu moczowego – kliniczne rozpoznanie zespołu Townesa-Brocksa. Przegląd Pediatryczny 2010; 40 (2): 122-124. 8. Faguer S, Pillet A, Chassaing N et al.: Nephropathy in Townes-Brocks syndrome (SALL1 mutation): imaging and pathological findings in adulthood. Nephrol Dial Transplant 2009; 24(4): 1341-1345. 9. Kohlhase J, Taschner PE, Burfeind P et al.: Molecular analysis of SALL1 mutations in Townes-Brocks syndrome. Am J Hum Genet 1999; 64(2): 435-4345. 10. Kohlhase J, Liebers M, Backe J et al.: High incidence of the R276X SALL1 mutation in sporadic but not familial Townes–Brocks syndrome and report of the first familial case. J Med Genet 2003; 40(11): e127. 11. Sudo Y, Numakura C, Abe A et al.: Phenotypic variability in a family with Townes-Brocks syndrome. J Hum Genet 2010; 55(8): 550-551. 12. Devriendt K, Fryns JP, Lemmens F et al.: Somatic mosaicism and variable expression of Townes-Brocks syndrome. Am J Med Genet 2002; 111(2): 230-231.