В© Borgis - PostДҷpy Nauk Medycznych 7/2014, s. 453-460
*Mariusz Marciszek, Emilia Wojtera
Receptory β-adrenergiczne w sercu (na marginesie nagrody nobla z chemii w 2012 roku)
Cardiac β-adrenoreceptors (reference to the nobel prize in chemistry in 2012 year)
ZakЕӮad Fizjologii Klinicznej, Centrum Medyczne KsztaЕӮcenia Podyplomowego, Warszawa
Kierownik ZakЕӮadu: prof. dr hab. med. Andrzej BerДҷsewicz
Streszczenie
W 2012 roku Kobilka i Lefkowitz otrzymali nagrodДҷ Nobla za caЕӮoksztaЕӮt prac nad receptorami sprzДҷЕјonymi z biaЕӮkiem G (GPCR). GPCR stanowiД… duЕјД… rodzinДҷ receptorГіw poЕӣredniczД…cych w przekazywaniu sygnaЕӮu ze Еӣrodowiska zewnДҷtrznego do wnДҷtrza komГіrki. CharakterystycznД… cechД… ich struktury jest siedem helis przenikajД…cych bЕӮonДҷ komГіrkowД…. ЕҒД…czy je takЕјe ogГіlny mechanizm transdukcji sygnaЕӮu. Polega on na zwiД…zaniu liganda w domenie zewnД…trzkomГіrkowej receptora i przeniesieniu pobudzenia poprzez zmiany konformacyjne na biaЕӮko G. Kaskada uruchamiana przez aktywacjДҷ receptora powoduje zmianДҷ metabolizmu komГіrki, wpЕӮywajД…c na aktywnoЕӣДҮ kinaz biaЕӮkowych i kanaЕӮГіw jonowych. Do tej rodziny naleЕјД… receptory β-AR obecne w sercu. Ich gЕӮГіwnД… rolД… w organizmie jest poЕӣredniczenie w natychmiastowej regulacji ukЕӮadu krД…Ејenia w odpowiedzi na bodЕәce stresowe. PrzewlekЕӮa stymulacja tych receptorГіw prowadzi do uaktywnienia ЕӣcieЕјek proprzerostowych w sercu. NiewydolnoЕӣДҮ serca (NS) charakteryzuje siДҷ przewlekЕӮД… nadmiernД… stymulacjД… receptorГіw β-AR. Skutkuje to zmianД… w skЕӮadzie iloЕӣciowym receptorГіw β-AR w sercu i niesie za sobД… zmianДҷ w odpowiedzi serca na sytuacje stresowe. Poznanie struktury i sposobu funkcjonowania receptorГіw β-AR pozwoliЕӮo na wprowadzenie odpowiedniej terapii u osГіb dotkniДҷtych NS. ArtykuЕӮ podsumowuje aktualny stan wiedzy dotyczД…cy budowy i funkcjonowania receptorГіw β-AR w sercu.
Summary
In 2012 Kobilka and Lefkowitz won the Nobel Prize in chemistry for their work on G-protein coupled receptors (GPCR). GPCRs comprise a large receptor family engaged in signal transduction from the external environment to the cell interior. The most characteristic feature of this family is a receptor structure that involves seven transmembrane helices arranged into a funnel. GPCRs have also common mode of signal transduction which consist in receptor rearrangement, caused by binded ligand, and as a result activation of G protein. The cascade started by receptor activation leads to changes in cellular metabolism through acting with protein kinases or ionic channels. Cardiac β-adrenergic receptors (β-AR) are the member of this family. Their main role is in immediate adaptation of the cardiovascular system to the external stressors. Heart failure (HF) is associated with chronic excessive β-AR stimulation that results in hypertrophic myocardial response and in quantitative changes in the cardiac β-AR expression and activity. Knowledge of the β-AR’s structure and mechanism of action set the stage for the contemporary HF therapy with β-AR antagonists.

WPROWADZENIE
W 2012 roku NagrodДҷ Nobla w dziedzinie chemii otrzymali Brian K. Kobilka i Robert J. Lefkowitz. Autorzy ci od lat 70. XX w. pracowali nad izolacjД…, krystalizacjД… i badaniem struktury receptorГіw β-adrenergicznych. Nagroda zostaЕӮa przyznana za caЕӮoksztaЕӮt prac obydwu naukowcГіw, ktГіre pozwoliЕӮy zrozumieДҮ, w jaki sposГіb funkcjonujД… i jak sД… zbudowane receptory sprzДҷЕјone z biaЕӮkiem G (ang. G-protein coupled receptor – GPCR), w tym β-AR. Receptory GPCR sД… duЕјД… rodzinД… receptorГіw bЕӮonowych, ktГіre: majД… podobnД… strukturДҷ, odbierajД… sygnaЕӮy chemiczne z otoczenia komГіrek niesione przez hormony, neurotransmitery, neuropeptydy, substancje parakrynne, substancje odЕјywcze, zapachy, smaki oraz poЕӣredniczД… w przekazywaniu tych sygnaЕӮГіw do wnДҷtrza komГіrki za poЕӣrednictwem wewnД…trzkomГіrkowego efektora, jakim jest biaЕӮko G (1, 2).
W sercu obecne sД… receptory adrenergiczne β1, β2 i β3 (β-AR) rГіЕјniД…ce siДҷ powinowactwem do agonistГіw, komГіrkowymi szlakami sygnalizacyjnymi i funkcjД… biologicznД…. Stosunek gДҷstoЕӣci β1-AR do β2-AR na powierzchni kardiomiocytГіw wynosi 3:1. W natychmiastowej regulacji ukЕӮadu krД…Ејenia (minuty), ktГіrej istota polega miДҷdzy innymi na zmianie czynnoЕӣci serca, udziaЕӮ biorД… gЕӮГіwnie β1-AR poprzez aktywacjДҷ szlaku kinazy biaЕӮkowej A. Wynika to z iloЕӣciowej przewagi β1-AR nad β2-AR oraz faktu, Ејe β1-AR majД… wiДҷksze powinowactwo do agonistГіw niЕј pozostaЕӮe β-AR. Wypadkowym efektem krГіtkotrwaЕӮej stymulacji wspГіЕӮczulnej serca sД…: wzrost siЕӮy skurczu miokardium, przyspieszenie rozkurczu, przyspieszenie czДҷstoЕӣci rytmu zatokowego i zwiДҷkszenie szybkoЕӣci przewodzenia w wДҷЕәle AV. Aktywacja β2-AR i β3-AR sЕӮuЕјy gЕӮГіwnie jako mechanizm ograniczajД…cy skutki nadmiernej aktywacji β1-AR, poprzez desensytyzacjДҷ i tzw. down-regulacjДҷ β1-AR. PrzedЕӮuЕјajД…ca siДҷ aktywacja wspГіЕӮczulna skutkuje aktywacjД… dodatkowych ЕӣcieЕјek sygnalizacyjnych prowadzД…cych do przerostu miДҷЕӣnia sercowego. W przypadku regularnego treningu sportowego aktywowane sД… szlaki skutkujД…ce blokowaniem apoptozy i tzw. przerostem fizjologicznym (poprzez aktywacjДҷ β2-AR i szlaki „pro-life”), a w przypadku niewydolnoЕӣci krД…Ејenia (NS) dodatkowo szlaki skutkujД…ce przyspieszonД… apoptozД… i tzw. przerostem patologicznym (poprzez aktywacjДҷ β1-AR i szlaki „pro-death”). W NS ekspresja i aktywnoЕӣДҮ β1-AR w sercu sД… obniЕјone, co z jednej strony chroni je przed nadmiernД… stymulacjД… wspГіЕӮczulnД…, a z drugiej – ogranicza moЕјliwoЕӣДҮ wzrostu rzutu minutowego serca podczas wysiЕӮku i w rГіЕјnych sytuacjach stresowych.
STRUKTURA RECEPTORГ“W GPCR
W ludzkim organizmie wykryto kilkaset receptorГіw GPCR. BiaЕӮka te sД… zbudowane z pojedynczego ЕӮaЕ„cucha polipeptydowego o pofaЕӮdowanej strukturze, ktГіry siedmiokrotnie przenika bЕӮonДҷ komГіrkowД…, tworzД…c w jej rdzeniu domenДҷ hydrofobowД… oraz pozabЕӮonowe domeny: zewnД…trz- i wewnД…trzkomГіrkowД…. Domena wewnД…trzbЕӮonowa zbudowana jest z α-helis (oznaczonych symbolami TM1-TM7, liczД…c od N-koЕ„ca), ktГіrych wzajemne uЕӮoЕјenie tworzy okrД…gЕӮy lub lejkowaty ksztaЕӮt. Helisy poЕӮД…czone sД… ze sobД… pДҷtlami wystajД…cymi ponad bЕӮonДҷ – do cytozolu i na zewnД…trz komГіrki. Domena wewnД…trzkomГіrkowa receptora tworzona jest przez pДҷtle ICL1-ICL3 (ang. intracellular loop) eksponowane do wnДҷtrza komГіrki oraz C-koniec ЕӮaЕ„cucha polipeptydowego. PДҷtla ICL3, ЕӮД…czД…ca helisy bЕӮonowe TM5 z TM6, wspГіlnie z pДҷtlД… ICL2 oraz fragmentem C-koЕ„ca tworzy miejsce wiД…zania i aktywacji biaЕӮka G. Domena zewnД…trzkomГіrkowa tworzona jest przez pДҷtle ECL1-ECL3 znajdujД…ce siДҷ poza cytozolem (ang. extracellular loop) oraz przez N-koniec biaЕӮka. W czДҷЕӣci zewnД…trzkomГіrkowej receptora zachodzi wiД…zanie ligandГіw. WejЕӣcie dla nich znajduje siДҷ pomiДҷdzy pДҷtlД… ECL3 a poЕӮД…czonymi pДҷtlami ECL1 i ECL2 (ktГіre tworzД… przykrywkДҷ nad miejscem wiД…zania ligandu). Budowa domeny zewnД…trzkomГіrkowej moЕјe rГіЕјniДҮ siДҷ w szczegГіЕӮach pomiДҷdzy rГіЕјnymi receptorami, jednak ogГіlny schemat jest wspГіlny (1, 3-5).
CzДҷЕӣci wewnД…trz- i zewnД…trzkomГіrkowa receptora mogД… ulegaДҮ modyfikacjom potranslacyjnym. W czДҷЕӣci zewnД…trzkomГіrkowej mogД… powstawaДҮ mostki siarczkowe stabilizujД…ce strukturДҷ przestrzennД… receptora. W β-AR mostki spinajД…ce pДҷtlДҷ ECL3 otwierajД… wiДҷkszy dostДҷp ligandГіw do kieszeni receptora. CzДҷЕӣДҮ wewnД…trzkomГіrkowa moЕјe ulegaДҮ glikozylacji, mirystylacji i palmitynizacji lub moЕјe przyЕӮД…czaДҮ czД…steczki cholesterolu. Modyfikacje te majД… znaczenie w kierowaniu receptora do miejsc w bЕӮonie komГіrkowej bogatej w kaweole lub tratwy lipidowe. Dodatkowo, okolice C-koЕ„ca bogate w serynДҷ i treoninДҷ mogД… byДҮ fosforyzowane przez odpowiednie kinazy, co skutkuje inaktywacjД… receptora (vide poniЕјej) (4).
MECHANIZM TRANSDUKCJI SYGNAЕҒU GPCR
Po przyЕӮД…czeniu siДҷ liganda do receptora nastДҷpuje zmiana konformacyjna caЕӮego receptora. Polega to na takiej zmianie uЕӮoЕјenia przestrzennego helis transbЕӮonowych, Ејe moЕјliwe staje siДҷ zwiД…zanie biaЕӮka G z wewnДҷtrznД… domenД… receptora. NastДҷpuje aktywacja biaЕӮka G, ktГіra skutkuje zmianД… aktywnoЕӣci wewnД…trzkomГіrkowego biaЕӮka efektorowego (najczДҷЕӣciej kinazy biaЕӮkowej lub kanaЕӮu jonowego) i zmiana komГіrkowego poziomu substancji peЕӮniД…cej funkcjДҷ przekaЕәnika informacji drugiego rzДҷdu (np. cAMP). Jednorazowa aktywacja receptora skutkuje aktywacjД… wielu kopii biaЕӮka G, co powoduje wzmocnienie sygnaЕӮu. TransmisjДҷ sygnaЕӮu blokuje fosforylacja receptora przez odpowiednie kinazy (vide poniЕјej) (3, 4, 6, 7).
BiaЕӮko G jest heterotrimerem i skЕӮada siДҷ z podjednostek α, β i γ (istniejД… takЕјe biaЕӮka G0 o budowie monomerycznej). Podjednostka α wiД…Ејe nukleotyd guanylowy (GTP) i ma aktywnoЕӣДҮ GTP-azy. Natomiast β i γ tworzД… niepodzielny kompleks, ktГіry ЕӮД…czy siДҷ z nieaktywnД… podjednostkД… α. Zaktywowane biaЕӮko G zmienia konformacjДҷ i czД…steczka GDP zwiД…zana z podjednostkД… α jest wymieniana na GTP z cytozolu, co umoЕјliwia odЕӮД…czenie podjednostki α od kompleksu βγ. BiaЕӮko G podzielone na komponenty α i βγ przekazuje sygnaЕӮ na biaЕӮko efektorowe (np. cyklazДҷ adenylowД…), czemu towarzyszy hydroliza GTP do GDP i nieaktywna podjednostka α odnawia wiД…zanie z kompleksem βγ (5, 7-9).
Istnieje kilka izoform biaЕӮka G (Gs, Gi, Gt, Gq(p), G0) rГіЕјniД…cych siДҷ biaЕӮkiem efektorowym, z ktГіrym wchodzД… w interakcjДҷ. BiaЕӮko Gs aktywuje cyklazДҷ adenylowД…, ktГіra produkuje cAMP (7-10). Gi hamuje cyklazДҷ adenylowД… i zmniejsza produkcjДҷ cAMP (7-10). Gq(p) aktywuje fosfolipazДҷ C, ktГіrej produktami sД… diacyloglicerol (DAG) i trifosforan inozytolu (IP3). Gt aktywuje fosfodiesterazДҷ, a biaЕӮko G0 hamuje kanaЕӮy wapniowe (7, 9).
Obok podjednostki α takЕјe kompleks βγ ma dziaЕӮanie sygnalizacyjne. Aktywuje mianowicie kinazДҷ receptorГіw GPCR (GRK), ktГіra fosforylujД…c biaЕӮko receptorowe, skutkuje zahamowaniem aktywnoЕӣci receptora, co jest mechanizmem zabezpieczajД…cym przed nadmiernД… aktywacjД… receptora (vide poniЕјej) (7, 11). Kompleks βγ bierze rГіwnieЕј udziaЕӮ w receptorowej aktywacji kanaЕӮГіw wapniowych i niektГіrych potasowych (7-9).
WPЕҒYW LIGANDГ“W NA AKTYWNOЕҡДҶ RECEPTORA
Receptory GPCR sД… czДҷsto przedstawiane w ujДҷciu bimodalnym. ZakЕӮada ono, Ејe receptor moЕјe wystДҷpowaДҮ tylko w jednym z dwГіch stanГіw – moЕјe byДҮ w stanie aktywnym, w ktГіrym miejsce wiД…zania biaЕӮka G jest odsЕӮoniДҷte, albo w stanie nieaktywnym, w ktГіrym miejsce wiД…zania biaЕӮka G jest zakryte. Wbrew tej koncepcji, niektГіre receptory GPCR, w tym β-AR, wykazujД… pewnД… podstawowД… aktywnoЕӣДҮ nawet pod nieobecnoЕӣДҮ agonisty. Okazuje siДҷ ponadto, Ејe podstawowa aktywnoЕӣДҮ receptorГіw moЕјe byДҮ regulowana przez ligandy. W tym kontekЕӣcie wyrГіЕјnia siДҷ: a) peЕӮnych agonistГіw – czyli ligandy skutkujД…ce maksymalnД… aktywacjД… receptora, b) czДҷЕӣciowych agonistГіw – czyli ligandy, ktГіre nawet w stДҷЕјeniach wysycajД…cych receptor skutkujД… jedynie submaksymalnД… aktywacjДҷ biaЕӮka G oraz c) odwrotnych agonistГіw – czyli ligandy, ktГіre hamujД… podstawowД… aktywnoЕӣДҮ receptora. Natomiast antagoniЕӣci nie modyfikujД… aktywnoЕӣci samego receptora, a jedynie kompetencyjnie blokujД… wiД…zanie z nim innych ligandГіw (2, 4, 12). Interakcja liganda z receptorem moЕјe odbywaДҮ siДҷ na dwa sposoby. Agonista moЕјe skutkowaДҮ zmianД… konformacji receptora poprzez proste zakЕӮГіcenie oddziaЕӮywaЕ„ istniejД…cych wewnД…trz jego czД…steczki i kreujД…c zestaw oddziaЕӮywaЕ„ stabilizujД…cych nowy stan konformacyjny lub teЕј moЕјe sЕӮuЕјyДҮ jako mostek, ktГіry bierze udziaЕӮ w powstawaniu nowych interakcji pomiДҷdzy domenami transbЕӮonowymi i stabilizuje bardziej aktywnД… konformacjДҷ receptora (2, 12).
SERCOWE EFEKTY NATYCHMIASTOWEJ AKTYWACJI β-AR
W sercu obecne sД… receptory adrenergiczne β1, β2 i β3 (β-AR). Agonistami β-AR w sercu sД… mediatory ukЕӮadu wspГіЕӮczulnego: noradrenalina – uwalniana z zakoЕ„czeЕ„ nerwowych sercowych wЕӮГіkien wspГіЕӮczulnych, oraz adrenalina – uwalniana z rdzenia nadnerczy. W zdrowym sercu stosunek gДҷstoЕӣci β1-AR do β2-AR wynosi 70-80%/20-30%. Dodatkowo, najwiДҷksze powinowactwo do agonistГіw majД… β1-AR, a najmniejsze β3-AR (okoЕӮo 100 razy mniejsze). Razem wziД…wszy, oznacza to, Ејe przy umiarkowanym poziomie aktywacji wspГіЕӮczulnej pobudzeniu ulegajД… gЕӮГіwnie β1-AR i Ејe dwa pozostaЕӮe β-AR wЕӮД…czajД… siДҷ dopiero w stanach znacznie zwiДҷkszonej aktywacji wspГіЕӮczulnej.
Receptory β-adrenergiczne sД… (ryc. 1):
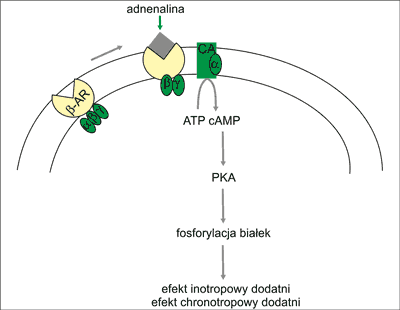
Ryc. 1. Schemat przekazywania sygnaЕӮu przez receptory β-ad-renergiczne.
β-AR – receptor adrenergiczny β; α, β, γ – podjednostki biaЕӮka G; CA – cyklaza adenylowa; ATP – adenozyno-trifosforan; cAMP – cykliczny adenozyno-monofosforan; PKA – kinaza biaЕӮkowa A
1. SprzДҷЕјone z biaЕӮkiem Gs (heterotrimer αsβγ) i/lub biaЕӮkiem Gi (heterotrimer αiβγ); podjednostka αs aktywuje cyklazДҷ adenylowД… i produkcjДҷ cAMP, a podjednostka αi enzym ten hamuje.
2. BiaЕӮkiem efektorowym szlaku β-AR w sercu sД…: a) enzym cyklaza adenylowa, ktГіry przeksztaЕӮca ATP w cykliczny adenozyno-mono-fosforan (cAMP) oraz b) kanaЕӮ wapniowy typu L.
3. DrugorzДҷdowymi przekaЕәnikami w ЕӣcieЕјce sygnalizacyjnej β-AR w sercu sД… cAMP oraz jony Ca2+.
4. Kinazami aktywowanymi przez ЕӣcieЕјkДҷ β-AR sД…: a) kinaza biaЕӮkowa A (PKA) aktywowana przez cAMP; PKA przenosi resztДҷ fosforanowД… z ATP na serynДҷ, treoninДҷ lub tyrozynДҷ rГіЕјnych biaЕӮek i w ten sposГіb zmienia ich wЕӮaЕӣciwoЕӣci, w tym wЕӮaЕӣciwoЕӣci biaЕӮek zaangaЕјowanych w skurcz bД…dЕә rozkurcz kardiomiocytГіw, czДҷstoЕӣДҮ pobudzeЕ„ wytwarzanych w wДҷЕәle zatokowo-przedsionkowym, szybkoЕӣДҮ przewodzenia w ЕӮД…czu AV oraz metabolizm komГіrki (ryc. 2) oraz b) kinaza aktywowana przez kompleks Ca2+-kalmodulina (CaMKII). Kinaza ta fosforyluje niektГіre biaЕӮka komГіrkowego obiegu jonГіw Ca2+ i w ten sposГіb uczestniczy w natychmiastowej regulacji pracy serca. Aktywuje takЕјe proprzerostowy szlak kalcyneuryny (ryc. 3).
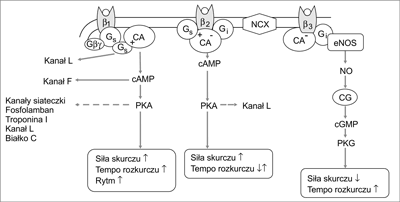
Ryc. 2. Szlaki aktywowane stymulacjД… typГіw receptorГіw adrenergicznych β1, β2 i β3 (objaЕӣnienia skrГіtГіw w tekЕӣcie).
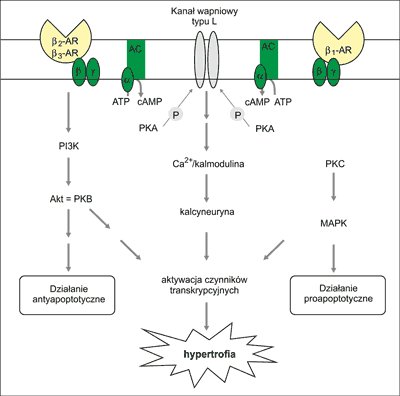
Ryc. 3. Mechanizmy zabezpieczajД…ce komГіrkДҷ przed nadmiernД… stymulacjД… katecholaminowД…. Sekwencja wydarzeЕ„ obejmuje: (1) aktywacjДҷ β-AR; (2) fosforylacjДҷ β-AR przez β-ARK; (3) blokowanie interakcji β-AR-biaЕӮko Gs przez β-arrestynДҷ; (4) internalizacjДҷ β-AR w mechanizmie ich endocytozy oraz (5) β-ARK zamkniДҷty w endosomie jest albo magazynowany w cytoplazmie, albo wraca na powierzchniДҷ bЕӮony, albo ulega degradacji w lizosomach.
Receptory β1
PowyЕјej zamieЕӣciliЕӣmy fragment artykuЕӮu, do ktГіrego moЕјesz uzyskaДҮ peЕӮny dostДҷp.
Mam kod dostДҷpu
- Aby uzyskaДҮ pЕӮatny dostДҷp do peЕӮnej treЕӣci powyЕјszego artykuЕӮu albo wszystkich artykuЕӮГіw (w zaleЕјnoЕӣci od wybranej opcji), naleЕјy wprowadziДҮ kod.
- WprowadzajД…c kod, akceptujД… PaЕ„stwo treЕӣДҮ Regulaminu oraz potwierdzajД… zapoznanie siДҷ z nim.
- Aby kupiДҮ kod proszДҷ skorzystaДҮ z jednej z poniЕјszych opcji.
Opcja #1
24 zЕӮ
Wybieram
- dostДҷp do tego artykuЕӮu
- dostДҷp na 7 dni
uzyskany kod musi byДҮ wprowadzony na stronie artykuЕӮu, do ktГіrego zostaЕӮ wykupiony
Opcja #2
59 zЕӮ
Wybieram
- dostДҷp do tego i pozostaЕӮych ponad 7000 artykuЕӮГіw
- dostДҷp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zЕӮ
Wybieram
- dostДҷp do tego i pozostaЕӮych ponad 7000 artykuЕӮГіw
- dostДҷp na 90 dni
- oszczДҷdzasz 28 zЕӮ
PiЕӣmiennictwo
1. Nygaard R, Frimurer TM, Holst B et al.: Ligand binding and micro-switches in 7TM receptor structures. Trends Pharmacol Sci 2009; 30: 249-259.
2. Kobilka BK, Deupi X: Conformational complexity of G-protein-coupled receptors. Trends Pharmacol Sci 2007; 28: 397-406.
3. Schertler G: Structure of rhodopsin and the metarhodopsin I photointermediate. Curr Opin Struct Biol 2005; 15: 408-415.
4. Kobilka BK, Schertler G: New G-protein-coupled receptor crystal structures: insights and limitations. Trends Pharmacol Sci 2008; 29: 79-83.
5. Kobilka BK: G protein coupled receptor structure and activation. Biochim Biophys Acta 2007; 1768: 794-807.
6. Soubias O, Gawrisch K: The role of the lipid matrix for structure and function of the GPCR rhodopsin. Biochim Biophys Acta 2012; 1818: 234-240.
7. Traczyk W, Trzebski A: Fizjologia czЕӮowieka z elementami fizjologii stosowanej i klinicznej. Wydawnictwo Lekarskie PZWL, Warszawa 2004.
8. Hoyer D, Bartfai T: Neuropeptides and Neuropeptide Receptors: Drug Targets, and Peptide and Non-Peptide Ligands: a tribute to Prof. Dieter Seebach. Chemistry & Biodiversity 2012; 9: 2367-2387.
9. Lefkowitz RJ: Historical review: A brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol Sci 2004; 25: 413-422.
10. Lefkowitz RJ: Heterogeneity of adenylate cyclase-coupled Гҹ-adrenergic receptors. Biochem Pharmacol 1975; 24: 583-590.
11. Whalen EJ, Foster MW, Matsumoto A et al.: Regulation of Гҹ Adrenergic Receptor Signaling by S-Nitrosylation of G-Protein-Coupled Receptor Kinase 2. Cell 2007; 129: 511-522.
12. Zocher M, Fung JJ, Kobilka BK: Ligand-Specific Interactions Modulate Kinetic, Energetic, and Mechanical Properties of the Human Гҹ2 Adrenergic Receptor. Structure 2012; 20: 1391-1402.
13. Mackiewicz U, Klemenska E, BerДҷsewicz A: Receptory beta-adrenergiczne w zdrowym i niewydolnym sercu. Kardiol Pol 2007; 65: 368-375.
14. Molenaar P, Parsonage WA: Fundamental considerations of beta-adrenoreceptor subtypes in human heart failure. Trends Pharmacol Sci 2005; 26: 368-375.
15. Zhu WZ, Zheng M, Koch WJ et al.: Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci U S A 2001; 98: 1607-1612.
16. Razeghi P, Young ME, Alcorn JL et al.: Metabolic Gene Expression in Fetal and Failing Human Heart. Circulation 2001; 104: 2923-2931.
17. Marks AR: Ryanodine receptors, FKBP12, and heart failure. Front Biosci 2002; 7: d970-d97.
18. Doi M, Yano M, Kobayashi S et al.: Propranolol prevents the development of heart failure by restoring FKBP12.6-mediated stabilization of ryanodine receptor. Circulation 2002; 105: 1374-1379.