© Borgis - Postępy Nauk Medycznych 7/2014, s. 470-478
*Urszula Mackiewicz
Rola zaburzeń równowagi współczulno-przywspółczulnej w powstawaniu zaburzeń rytmu serca
The role of sympathetic-parasympathetic balance in arrhythmogenesis
Zakład Fizjologii Klinicznej, Centrum Medyczne Kształcenia Podyplomowego, Warszawa
Kierownik Zakładu: prof. dr hab. med. Andrzej Beręsewicz
Streszczenie
Około połowa osób z chorobami układu krążenia umiera w sposób nagły z powodu arytmii. Zagrażające życiu arytmie powstają głównie w mechanizmie pobudzenia nawrotnego i często towarzyszy im przewlekła lub nagła aktywacja układu współczulnego. Arytmia nawrotna występuje wtedy, gdy czynnik wyzwalający arytmię napotka na sprzyjające warunki elektrofizjologiczne. Czynnikiem wyzwalającym arytmie nawrotne są przedwczesne pobudzenia dodatkowe, powstające w mechanizmie wczesnych i późnych depolaryzacji następczych oraz automatyzm patologiczny komórek roboczych lub komórek Purkinjego. Substratem elektrofizjologicznym do powstania arytmii jest istnienie dyspersji refrakcji w sąsiadujących ze sobą kardiomiocytach. Prowadzi to do powstania jednokierunkowego bloku przewodzenia, który jest warunkiem koniecznym do zawiązania się pętli pobudzenia nawrotnego. Dodatkowo zawiązaniu się i podtrzymaniu pętli sprzyjają wolne przewodzenie pobudzenia i krótkie okresy refrakcji. Katecholaminy zwiększają zarówno częstość pobudzeń dodatkowych (głównie powstających w mechanizmie późnych depolaryzacji następczych), jak i nasilają substrat elektrofizjologiczny, zwiększając dyspersję refrakcji. Proarytmiczne działanie katecholamin jest szczególnie widoczne w takich stanach patologicznych jak niewydolność serca, zawał serca czy zaburzenia elektrolitowe. β-blokery redukują częstość arytmii i liczbę nagłych zgonów sercowych, przeciwdziałając proarytmicznym komórkowym efektom stymulacji katecholaminowej.
Summary
About half of patient with cardiovascular diseases die suddenly due to arrhythmic reasons. A chronic or immediate activation of the sympathetic nervous system often accompanies life threatening arrhythmias (predominantly of reentrant nature). The reentrant arrhythmias are induced, when a pro-arrhythmic trigger meets a adequate electrophysiological substrate. Premature ventricular ectopic beats arising from early or delayed afterdepolariztions as well as from abnormal automaticity in Purkinje fibers or ventricular cardiomyocytes are the main triggers of reentrant arrhythmias. In turn, dispersion of refractoriness (necessary condition for unidirectional block of conduction) is the main electrophysiological substrate for initiation of reentrant circuit. Additionally, slow impulse propagation and short refractory period permit maintenance of reentry. Catecholamines increase both frequency of the premature beats (especially arising from delayed afterdepolarizations) and electrophysiological substrate (heterogeneity of refractoriness). Proarrhythmic catecholamine effects are especially significant in pathological states such as the heart failure, myocardial infarction and electrolyte imbalance. β-blockers reducing proarrhythmic action of catecholamines prevent arrhythmias and sudden cardiac deaths.

ZWIĄZEK WYSTĘPOWANIA ARYTMII Z AKTYWACJĄ UKŁADU WSPÓŁCZULNEGO
Najgroźniejszym następstwem zaburzeń rytmu serca jest nagły zgon sercowy (NZS). Najczęściej (ponad 90% przypadków) NZS ma podłoże arytmiczne. W sposób nagły, w następstwie arytmii, umiera ok. 50% pacjentów z chorobami układu krążenia. Pojawienie się zagrażających życiu arytmii i NZS udaje się powiązać z nagłą lub przewlekłą aktywacją układu współczulnego. Leki znoszące komórkowe następstwa stymulacji katecholaminowej (β-blokery) redukują liczbę NZS (1).
Najczęstszą przyczynę NZS (ponad 80%) stanowi choroba niedokrwienna serca i jej najgroźniejsza manifestacja – ostry zespół wieńcowy. Śmiertelność w zawale serca w dobie inwazyjnego leczenia choroby wieńcowej spada i jest szacowana na ok. 5%. Z tego około 40-50% umiera w pierwszej dobie zawału serca, większość przed uzyskaniem pomocy szpitalnej. Kolejne 3-20% umiera w ciągu 2-3 tygodni po zawale (połowa z nich w sposób nagły). Łącznie szacuje się, że około połowa zgonów w zawale serca spowodowana jest groźnymi arytmiami komorowymi (2, 3).
W zawale serca spadek ciśnienia krwi i niedostateczna perfuzja narządowa oraz ból i stres istotnie zwiększają aktywację układu współczulnego (4). β-blokery skutecznie redukują ilość NZS w zawale (5, 6). Metaanaliza licznych badań wpływu β-blokerów na śmiertelność po zawale serca, obejmująca ponad 50 000 pacjentów, potwierdziła wysoką skuteczność tych leków w prewencji wtórnej. Średnia redukcja śmiertelności wynosiła 23% (7). Ponadto wykazano, że dołączenie β-blokera do terapii inhibitorem konwertazy angiotensyny (ACE-inhibitor) skutecznie obniżało śmiertelność po zawale (8).
Druga grupa chorych ze szczególnie wysokim odsetkiem NZS to pacjenci z niewydolnością serca. Podobnie jak w zawale serca, około 50% chorych umiera w mechanizmie NZS z przyczyn arytmicznych (9). Aktywacja neurohumoralna, obejmująca aktywację układu współczulnego i układu renina-angiotensyna-aldosteron (RAAS), jest głównym patomechanizmem tej choroby. Dodatkowo obserwuje się spadek aktywności układu przywspółczulnego. Stężenie noradrenaliny w tkance mięśnia sercowego rośnie istotnie już w mało zaawansowanym, a nawet bezobjawowym stadium choroby, a w zaawansowanej niewydolności serca odnotowuje się nawet 50-krotny wzrost stężenia tego hormonu (porównywalny do tego, jaki obserwuje się u zdrowych osób po maksymalnym wysiłku). Komórki niewydolnego serca są więc stale eksponowane na bardzo wysokie stężenie katecholamin (10, 11). Skuteczność β-blokerów w zapobieganiu NZS w niewydolności serca potwierdziły liczne badania wieloośrodkowe. Pokazano, że uzupełnienie terapii obejmującej ACE-inhibitory i diuretyki przez β-bloker redukowało częstość występowania nagłych zgonów sercowych. Ogólna redukcja nagłych zgonów na podstawie metaanalizy obejmującej 6 największych badań wieloośrodkowych przeprowadzonych wśród pacjentów z niewydolnością serca wynosiła 38% (12). Wykazano również, że u pacjentów ze stabilną niewydolnością serca rozpoczęcie terapii od β-blokera i następnie dołączenie ACE-inhibitora silniej redukowało częstość NZS niż rozpoczęcie leczenia w odwrotnej konwencji (13).
Odrębną grupę NZS stanowią zgony osób młodych (przed 35. rokiem życia) bez objawów ze strony układu krążenia. Są to najczęściej osoby, u których arytmia ma podłoże genetyczne. Defekty dotyczą genów odpowiedzialnych za ekspresję kanałów jonowych (zespół długiego QT-LQTS, zespół krótkiego QT-SQTS, zespół Brugadów), białek wewnątrzkomórkowego obiegu Ca2+ (zależny od katecholamin wielokształtny częstoskurcz komorowy – CPVT), czy też białek aparatu kurczliwego i cytoszkieletu komórkowego (arytmogenna dysplazja prawej komory, rodzinnie uwarunkowana kardiomiopatia przerostowa i rozstrzeniowa) (14). Epizody arytmii u tych chorych również udaje się powiązać ze wzrostem aktywacji układu współczulnego podczas wysiłku lub stresu. Dotyczy to przede wszystkim pacjentów z zespołem długiego QT (LQT) typu 1 i 2 oraz CPVT (15). Leki blokujące receptory adrenergiczne wykazały skuteczność w zapobieganiu arytmiom w tych zespołach (u pacjentów z LQT1 redukcja nagłych zgonów wynosiła 80%, a z LQT2 sięgała 60%) (16).
Powyżej przedstawione fakty sugerują, że nadmierna stymulacja katecholaminowa może być jedną z przyczyn niebezpiecznych arytmii i NZS. Do zrozumienia roli aktywacji układu współczulnego w inicjowaniu arytmii, i tym samym mechanizmu antyarytmicznego działania β-blokerów, niezbędne jest zrozumienie skomplikowanych komórkowych mechanizmów wywołujących i podtrzymujących arytmię w mięśniu sercowym.
MECHANIZM POWSTAWANIA NIEBEZPIECZNYCH ZABURZEŃ RYTMU. POBUDZENIE NAWROTNE
W około 90% przypadków bezpośrednią przyczyną zatrzymania krążenia i NZS jest migotanie komór i/lub częstoskurcz komorowy, znacznie rzadziej (w około 10% przypadków) bradyarytmia przechodząca w asystolię (3). Najczęstszą przyczyną częstoskurczu komorowego, mogącego przejść w migotanie komór, jest pobudzenie nawrotne (ang. reentry). Do arytmii w mechanizmie pobudzenia nawrotnego dochodzi, gdy w mięśniu sercowym współistnieją ze sobą odpowiedni substrat elektrofizjologiczny (nierówne okresy refrakcji sąsiadujących ze sobą komórek i wolne przewodzenie pobudzenia) oraz czynnik wyzwalający arytmię (pobudzenie dodatkowe, będące następstwem depolaryzacji następczych lub automatyzmu patologicznego) (ryc. 1A) (17).
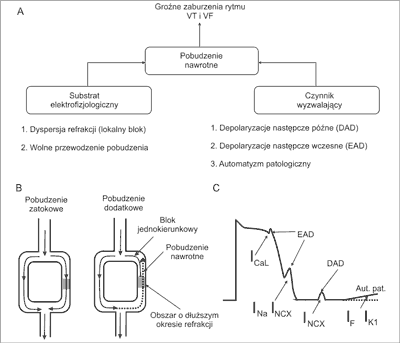
Ryc. 1. Mechanizm powstawania pobudzenia nawrotnego. (A) Czynniki predysponujące do powstania arytmii w mechanizmie pobudzenia nawrotnego; (B) blok jednokierunkowy i zawiązanie się pętli pobudzenia nawrotnego; (C) czynniki wyzwalające pobudzenie nawrotne i ich podłoże jonowe.
VT – częstoskurcz komorowy; VF – migotanie komór
Substrat elektrofizjologiczny
Warunkiem koniecznym do powstania pętli pobudzenia nawrotnego jest lokalny blok przewodzenia pobudzenia. Pobudzenie jest generowane w komórkach węzła zatokowo-przedsionkowego. Następnie obejmuje tkankę przedsionków, aktywując ich skurcz, i dociera do węzła przedsionkowo-komorowego. Stąd poprzez komorowy układ bodźco-przewodzący dociera do zlokalizowanych pod wsierdziem komórek roboczych (kardiomiocytów) lewej i prawej komory serca, rozchodząc się dalej w kierunkach od wsierdzia do nasierdzia i od koniuszka do podstawy komór. Tor przewodzenia w tkance roboczej komór serca jest skomplikowany. Obliczono, że średnio jeden kardiomiocyt ma styczność z jedenastoma innymi kardiomiocytami. Przewodzenie odbywa się z większą szybkością wzdłuż długiej osi kardiomiocytów, ale jest możliwe również wzdłuż osi krótkiej. Niejednokrotnie tor przewodzenia rozgałęzia się i często ponownie po pokonaniu pewnego dystansu rozgałęzienia się łączą (ryc. 1B). Jeżeli kardiomiocyty w poszczególnych rozgałęzieniach mają odmienne okresy refrakcji, to może dojść do sytuacji, w której pobudzenie pobudzi kardiomiocyty leżące w obrębie jednego rozgałęzienia, a zostanie zablokowane w drugim, gdzie kardiomiocyty pozostają jeszcze w okresie refrakcji. W ten sposób powstaje lokalny blok przewodzenia. W wyniku wystąpienia lokalnego bloku, pobudzenie zostaje przewiedzione tylko w jednym rozgałęzieniu toru i gdy dotrze do miejsca, w którym rozgałęzienia ponownie się łączą, może wejść wstecznie na drogę, która była poprzednio w okresie refrakcji, a po czasie kiedy pobudzenie dotarło do połączenia obu ramion, już z niego wyszła (ryc. 1B, prawy panel). Dochodzi do zawiązania się pętli pobudzenia nawrotnego. Zaistnienie takiej sytuacji jest tym bardziej prawdopodobne, im wolniejsze jest przewodzenie pobudzenia, ponieważ droga o dłuższym okresie refrakcji zyskuje czas na powrót do pobudliwości i może zostać pobudzona wstecznie. Ponadto powstaniu pobudzenia nawrotnego sprzyjają względnie krótkie okresy refrakcji. Wtedy to droga, w której pierwotnie doszło do bloku, może być pobudzona wstecznie po stosunkowo krótkim czasie (18, 19).
Bodziec wyzwalający
Jeżeli serce jest pobudzane w rytmie zatokowym, szansa na zawiązanie się pętli pobudzenia nawrotnego jest niewielka. Czas, jaki upływa między pobudzeniami, jest na tyle długi, że przed kolejnym pobudzeniem zatokowym okres refrakcji zakończy się w obu rozgałęzieniach toru przewodzenia (nawet przy istnieniu istotnej dyspersji refrakcji) i pobudzenie zostanie przewiedzione w każdej z nich (ryc. 1B, lewy panel). Jeżeli jednak w mięśniu komór pojawi się dodatkowe przedwczesne pobudzenie komorowe, szansa na to, że w drodze o dłuższej refrakcji zostanie ono zablokowane i dojdzie do lokalnego bloku przewodzenia, jest duża (ryc. 1B, prawy panel).
Komorowe pobudzenia dodatkowe powstają w wyniku późnych (ang. Delayed After Depolarizations – DAD) i wczesnych (ang. Early After Depolarizations – EAD) depolaryzacji następczych, występujących odpowiednio po zakończeniu potencjału czynnościowego oraz w trakcie fazy plateau lub repolaryzacji potencjału czynnościowego. Dodatkowym źródłem pobudzeń dodatkowych może być również automatyzm patologiczny komórek roboczych lub komórek Purkinjego (ryc. 1C) (17-19).
Zawiązaniu się i podtrzymaniu arytmii w mechanizmie pobudzenia nawrotnego sprzyjają niehomogenne okresy refrakcji, wolne przewodzenie pobudzenia oraz przedwczesne pobudzenia dodatkowe.
JONOWE MECHANIZMY ARYTMII TYPU POBUDZENIA NAWROTNEGO I ICH REGULACJA PRZEZ KATECHOLAMINY
Dyspersja refrakcji
Dyspersja okresów refrakcji stwarza warunki do powstania lokalnego bloku przewodzenia i zawiązania się pętli pobudzenia typu nawrotnego (ryc. 1A i B). Okres refrakcji komórek zależy głównie od czasu trwania ich potencjałów czynnościowych, który z kolei jest determinowany przez wielkość depolaryzującego prądu wapniowego typu L (CaL) i repolaryzującego prądu potasowego typu Kr (szybki dokomórkowo-prostowniczy prąd z opóźnioną aktywacją) (20) (ryc. 2). W sytuacji kiedy dochodzi do wzrostu prądu CaL lub spadku prądu Kr, potencjał wydłuża się; podczas odwrotnych zmian ulega skróceniu. Ta prosta zależność odpowiedzialna za czas trwania potencjału w warunkach fizjologicznych może być w sytuacjach patologicznych komplikowana przez dodatkowe prądy. Ważnym uczestnikiem gry o czas trwania potencjału staje się wtedy prąd płynący przez kanał Ks (wolny kanał potasowy o opóźnionej aktywacji) stanowiący tzw. rezerwę repolaryzacji. Prąd ten w warunkach fizjologicznych bierze niewielki udział w repolaryzacji, ale jego udział rośnie bardzo istotnie podczas stymulacji katecholaminowej oraz w sytuacjach, gdy dochodzi do zmniejszenia funkcji kanału Kr (hipopotasemia, leki blokujące kanał Kr, defekty genetyczne). Dodatkowym prądem mogącym wpływać na czas trwania potencjału w warunkach patologicznych jest prąd sodowy (Na+). W komórkach niewydolnego serca dochodzi do opóźnionego zamykania kanałów Na+ (to zjawisko obserwuje się również u chorych z LQT typu 3) i prąd, który fizjologicznie bierze udział tylko w szybkiej depolaryzacji przedłuża się na fazę plateau i wydłuża potencjał (ryc. 2). Z kolei w obszarach niedokrwionego mięśnia może dochodzić do otwarcia kanałów potasowych regulowanych spadkiem poziomu ATP lub wzrostem stężenia Ca2+ i do skrócenia potencjału (17).
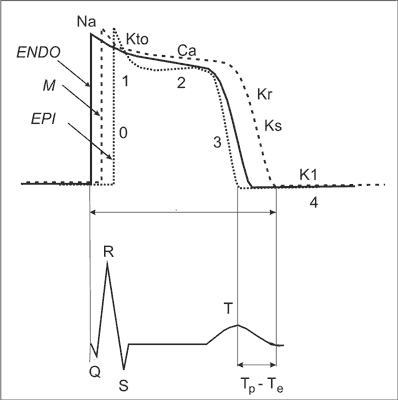
Ryc. 2. Śródścienna dyspersja repolaryzacji w mięśniu sercowym. EPI – komórki podnasierdziowe; M – komórki warstwy środkowej mięśnia sercowego; ENDO – komórki podwsierdziowe; 0 – faza szybkiej depolaryzacji; 1 – faza wstępnej repolaryzacji; 2 – faza plateau; 3 – faza szybkiej repolaryzacji; 4 – faza potencjału spoczynkowego
Czas trwania potencjałów w kardiomiocytach komór mięśnia sercowego jest różny i wynika ze zróżnicowanej gęstości kanałów jonowych w ich błonach komórkowych. Różnice w gęstości kanałów i czasie trwania potencjałów stwierdza się między komórkami lewej i prawej komory oraz między komórkami koniuszka i podstawy (tzw. dyspersja koniuszkowo-podstawna). Jednak zdecydowanie największe różnice występują w przekroju poprzecznym ściany mięśnia sercowego (tzw. dyspersja śródścienna). Najkrótsze potencjały rejestruje się w komórkach podnasierdziowych (EPI) i wynika to ze znamiennie większej w tym obszarze gęstości kanałów Kr niż w komórkach podwsierdziowych (ENDO), w których potencjały są dłuższe (ryc. 2). Dzięki takiej dystrybucji kanałów w poszczególnych obszarach mięśnia komórki EPI, które są pobudzane jako ostatnie, repolaryzują się jako pierwsze (depolaryzacja rozchodzi się od wsierdzia do nasierdzia, a repolaryzacja od nasierdzia do wsierdzia). Pomiędzy komórkami EPI i ENDO znajdują się jeszcze tzw. komórki M o najdłuższych potencjałach czynnościowych. Wydłużenie potencjału komórek M wynika z opóźnionego zamykania się ich kanałów Na+ (tzw. późny prąd sodowy) i dużego depolaryzującego prądu wymiennika Na+/Ca2+ (NCX). W tych komórkach stwierdza się również bardzo niską gęstość kanałów Ks w porównaniu z komórkami EPI i ENDO. W warunkach fizjologicznych nie ma to wielkiego wpływu na czas trwania potencjału komórek M, ale może go istotnie wydłużać w warunkach, w których potrzebne jest uruchomienie rezerwy repolaryzacji. Ostatecznie, komórki EPI depolaryzują się jako pierwsze, a komórki M jako ostatnie i różnica pomiędzy czasem trwania potencjałów w tych komórkach decyduje o śródściennej dyspersji repolaryzacji (ryc. 2). W zdrowym mięśniu dyspersja repolaryzacji jest niewielka. Dzieje się tak dlatego, że kanał Ks bierze niewielki udział w repolaryzacji w warunkach fizjologicznych. Dodatkowo, komórki ENDO są ściśle połączone elektrycznie z komórkami M i ich potencjały się homogenizują (21). Natomiast w sytuacjach patologicznych, takich jak niewydolność serca, niedokrwienie mięśnia sercowego, zaburzenia elektrolitowe (szczególnie hipopotasemia), przyjmowanie leków wydłużających QT, defekty genetyczne w kanałach jonowych, dochodzi do niehomogennego wydłużenia potencjałów. Potencjały komórek M wydłużają się w tych warunkach znacznie silniej niż potencjały pozostałych komórek, co istotnie zwiększa dyspersję repolaryzacji (22).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Zipes DP, Wellens HJJ: Sudden Cardiac Death. Circulation 1998; 98: 2334-2351.
2. Myerburg RJ, Castellanos A: Cardiac arrest and sudden death. [In:] Braunwald E (ed.): Heart Disease: A Textbook of Cardiovascular Medicine. WB Saunders, Philadelphia 1997: 742-779.
3. ACC/AHA/ESC Guidelines for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death – Executive Summary. Eur Heart J 2006; 27: 2099-2140.
4. Vaseghi M, Shivkumar K: The role of the autonomic nervous system in sudden cardiac death. Prog Cardiovasc Dis 2008; 50: 404-419.
5. Chadda K, Goldstein S, Byington R, Curb JD: Effect of propranolol after acute myocardial infarction in patients with congestive heart failure. Circulation 1986; 73: 503-510.
6. Kennedy HL, Brooks MM, Barker AH et al.: Beta-blocker therapy in the Cardiac Arrhythmia Suppression Trial. CAST Investigators. Am J Cardiol 1994; 74: 674-680.
7. McMurray J, Køber L, Robertson M et al.: Antiarrhythmic effect of carvedilol after acute myocardial infarction: Results of the Carvedilol Post-Infarct Survival Control in Left Ventricular Dysfunction (CAPRICORN) trial. J Am Coll Cardiol 2005; 45: 531-532.
8. Freemantle N, Cleland J, Young P et al.: Beta blockade after myocardial infarction: Systematic review and meta regression analysis. BMJ 1999; 318: 1730-1737.
9. Merrit HF Investigators: Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF). Lancet 1999; 353: 2001-2007.
10. Triposkiadis F, Karayannis G, Giamouzis G et al.: The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol 2009; 54: 1747-1762.
11. Francis GS, Benedict C, Johnstone DE et al.: Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. A substudy of the Studies of Left Ventricular Dysfunction (SOLVD). Circulation 1990; 82: 1724-1729.
12. Teerlink JR, Massie BM: Beta-adrenergic blocker mortality trials in congestive heart failure. Am J Cardiol 1999; 84: 94R-102R.
13. Krum H, van Veldhuisen DJ, Funck-Brentano C et al.: Effect on Mode of Death of Heart Failure Treatment Started with Bisoprolol Followed by Enalapril, Compared to the Opposite Order: Results of the Randomized CIBIS III Trial. Cardiovasc Ther 2011; 29: 89-98.
14. Shah M, Akar FG, Tomaselli GF: Molecular basic for arrhythmia. Circulation 2005; 112: 2517-2529.
15. Biernacka EK: Left cardiac sympathetic denervation in the management of resistant catecholaminergic arrhythmias. Post Kardiol Interw 2011; 1: 61-64.
16. Moss A, Zareba W, Hall W et al.: Effectiveness and limitations of betablocker therapy in congenital long-QT syndrome. Circulation 2002; 101: 616-623.
17. Nattel S, Maguy A, LeBouter S, Yeh YH: Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev 2007; 87: 425-456.
18. Rubart M, Zipes DP: Genesis of Cardiac Arrhythmias: Electrophysiological Considerations. [In:] Braunwald E (ed.): Heart Disease: A Textbook of Cardiovascular Medicine. WB Saunders, Philadelphia 2007: 727-761.
19. Hoffman BF, Rosen MR: Cellular Mechanisms for Cardiac Arrhythmias. Circ Res 1981; 49: 1-15.
20. Nerbonne JM, Kass RS: Molecular physiology of cardiac repolarization. Physiol Rev 2005; 85: 1205-1253.
21. Schram G, Pourrier M, Melnyk P, Nattel S: Differential distribution of cardiac ion channel expression as a basis for regional specialization in electrical function. Circ Res 2002 May 17; 90(9): 939-950.
22. Conratha CE, Opthofb T: Ventricular repolarization: An overview of (patho)physiology, sympathetic effects and genetic aspects. Prog Biophys Mol Biol 2006; 92(3): 269-307.
23. Patel C, Burke JF, Patel H et al.: Is there a significant transmural gradient in repolarization time in the intact heart? Cellular basis of the T wave: a century of controversy. Circ Arrhythm Electrophysiol 2009; 2(1): 80-88.
24. Franz MR, Bargheer K, Rafflenbeul W et al.: Monophasic action potential mapping in human subjects with normal electrocardiograms: direct evidence for the genesis of the T wave. Circulation 1987; 75(2): 379-386.
25. Fish JM, Brugada J, Antzelevitch C: Potential proarrhythmic effects of biventricular pacing. J Am Coll Cardiol 2005; 46(12): 2340-2347.
26. Shimizu M, Ino H, Okeie K et al.: T-peak to T-end interval may be a better predictor of high-risk patients with hypertrophic cardiomyopathy associated with a cardiac troponin I mutation than QT dispersion. Clin Cardiol 2002; 25(7): 335-339.
27. Watanabe N, Kobayashi Y, Tanno K et al.: Transmural dispersion of repolarization and ventricular tachyarrhythmias. J Electrocardiol 2004; 37(3): 191-200.
28. Frishman WH, Lazar EJ: Reduction of mortality, sudden death and non-fatal reinfarction with beta-adrenergic blockers in survivors of acute myocardial infarction: a new hypothesis regarding the cardioprotective action of beta-adrenergic blockade. Am J Cardiol 1990; 66: 66-70.
29. Eslami V, Safi M, Taherkhani M et al.: Evaluation of QT, QT dispersion, and T-wave peak to end time changes after primary percutaneous coronary intervention in patients presenting with acute ST-elevation myocardial infarction. J Invasive Cardiol 2013; 25(5): 232-234.
30. Piccirillo G, Moscucci F, D’Alessandro G et al.: Myocardial repolarization dispersion and autonomic nerve activity in a canine experimental acute myocardial infarction model. Heart Rhythm 2013; 11(1): 110-118.
31. Yan GX, Antzelevitch C: Cellular Basis for the Normal T Wave and the Electrocardiographic Manifestations of the Long-QT Syndrome. Circulation 1998; 98: 1928-1936.
32. Bonnar CE, Davie AP, Caruana L et al.: QT dispersion in patients with chronic heart failure: beta blockers are associated with a reduction in QT dispersion. Heart 1999; 81(3): 297-302.
33. Bonnar CE, MacFadyen RJ, Robson JM et al.: QT dispersion is related to autonomic tone in patients with stable chronic heart failure (abstract). Eur Heart J 1997; 18: 200.
34. Bonnar CE, Gillespie ND, MacFadyen RJ et al.: QT dispersion is significantly increased between 6am and 8am in heart failure patients – a possible role in sudden death? J Am Coll Cardiol 1997; 29 (suppl. A): 510A.
35. Grassi G, Cattaneo BM, Mancia G: Sympathetic nervous system. [In:] Poole-Wilson PA (ed.): Heart failure. Churchill Livingstone, New York 1997: 199-214.
36. Barr CS, Naas AA, Fenwick M et al.: Enalapril reduces QT dispersion in patients with mild congestive heart failure secondary to coronary artery disease. Am J Cardiol 1997; 79: 328-333.
37. Dean JW, Lab MJ: Arrhythmia in heart failure. Role of mechanically induced changes in electrophysiology. Lancet 1989; 1(8650): 1309-1312.
38. Janse MJ, Wit AL: Electrophysiological mechanisms of ventricular arrhythmias resulting from myocardial ischemia and infarction. Physiol Rev 1989; 69: 1049-1069.
39. Pu J, Boyden PA: Alterations of Na+ currents in myocytes from epicardial border zone of the infarcted heart. A possible ionic mechanism for reduced excitability and postrepolarization refractoriness. Circ Res 1997; 81: 110-119.
40. Peters NS: Myocardial gap junction organization in ischemia and infarction. Microsc Res Tech 1995: 31: 375-386.
41. Kitamura H, Ohnishi Y, Yoshida A et al.: Heterogeneous loss of connexin43 protein in nonischemic dilated cardiomyopathy with ventricular tachycardia. Cardiovasc Electrophysiol 2002; 13: 865-870.
42. Bers DM, Gua T: Calcium signaling in cardiac ventricular myocytes. Ann NY Acad Sci 2005: 1047: 86-98.
43. Antoons G, Sipido KR: Targeting calcium handling in arrhythmias. Europace 2008; 10: 1364-1369.
44. Lohse MJ, Engelhardt S, Eschenhagen T: What is the role of beta-adrenergic signaling in heart failure? Circ Res 2003; 93: 896-906.
45. Pflaumer A, Davis AM: Guidelines for the diagnosis and management of Catecholaminergic Polymorphic Ventricular Tachycardia. Heart Lung Circ 2012; 21: 96-100.
46. Yano M, Ikeda Y, Matsuzaki M: Altered intracellular Ca2+ handling in hart failure. J Clin Invest 2005; 115: 556-564.
47. Pogwizd SM, Schlotthauer K, Li L et al.: Arrhythmogenesis and contractile dysfunction in heart failure: roles of sodium-calcium exchange, inward rectifier potassium current, residual beta-adrenergic responsiveness. Circ Res 2001; 88: 1159-1167.
48. Sillitano F, Lonardo G, Zicha S et al.: Molecular basis of funny current (If) in normal and failing human heart. J Mol Cell Cardiol 2008; 45(2): 289-299.
49. Litwin SE, Zhang D, Bridge JH: Dyssynchronous Ca2+ sparks in myocytes from infarcted hearts. Circ Res 2000; 87: 1040-1047.
50. Waldo AL, Wit AL: Mechanisms of cardiac, arrhythmias and conduction disturbances. [In:] Fuster V, Alexsander RW, O’Rourke RA (eds.): Hurst’s The Heart. 11th ed., McGraw-Hill, Columbus, Ohio 2004: 787-816.
51. Roden DM: Taking the “idio” out of “idiosyncratic”: predicting torsades de pointes. Pacing Clin Electrophysiol 1998: 21: 1029-1034.
52. Krupa W, Kozłowski D: Genetyczne podstawy zaburzeń rytmu serca. Folia Cardiol 2000; 7(4): 273-279.
53. Peters NS, Cabo C, Wit AL: Arrhytmogenic mechanisms: Automaticity, triggered activity, and reentry. [In:] Zipes DP, Jalife J (eds.): Cardiac Electrophysiology: From Cell to Bedside. 3rd ed., WB Saunders, Philadelphia 2000: 345-355.
54. Mackiewicz U, Gerge JY, Chu S et al.: Ivabradine protects against ventricular arrhythmias in acute myocardial infarction in the rat. J Cell Physiol 2014, in press.