© Borgis - Medycyna Rodzinna 1/2004, s. 13-15
Sylwester Mordarski
Rola tlenku azotu w przewodnictwie neuronalnym
The role of nitric oxide in neurotransmission
z Katedry i Kliniki Anestezjologii i Intensywnej Terapii Akademii Medycznej we Wrocławiu
Kierownik Katedry i Kliniki: prof. dr hab. Andrzej Kübler
Summary
Nitric oxide (NO), a diffusible gas which nevertheless acts as a transmitter is produced in response to NMDA receptor activation and so may well contribute to some or all of the conesquences of NMDA receptor activation in nociception. There are blockers of nitric oxide synthase (NOS) which are, for example, effective against inflammatory and neuropathic nociception in animals by spinal actions. There will be problem with NOS blockers in therapy since NOS is endothelium - derived - relaxing factor and systemic administration of NOS blockers may induce analgesia but will be accompanied by severe hypertension. However, since it has been demonstrated that neuronal NOS differs from that in the endothelium, it should be possible to separate these effects.

Od ponad 100 lat anestezjolodzy używają podtlenku azotu (N2O), nieorganicznego gazu, jako środka anestetycznego. Ostatnio wiele uwagi poświęcono badaniom nad tlenkiem azotu pokrewnemu podtlenkowi azotu związkowi chemicznemu mającemu całkowicie różne właściwości chemiczne i fizjologiczne. Nowsze badania sugerują, że ta toksyczna gazowa molekuła może być endogennym neuronalnym przekaźnikem. Rola tlenku azotu w neurotransmisji i patologii centralnego układu nerwowego jest obecnie tematem intensywnych badań.
Tlenek azotu (NO) po raz pierwszy odkryto podczas badań nad wywodzącym się ze śródbłonka naczyniowego czynnikiem rozluźniającym mięśnie gładkie naczyń krwionośnych (endothelium – derived relaxing factor – EDRF) odpowiedzialnym za rozszerzanie naczyń krwionośnych w tkankach. Wiele z właściwości EDRF posiada również tlenek azotu. Należą do nich: 1) krótki czas półtrwania, 2) unieczynnianie przez hemoglobinę (inaktywacja hemoglobinowa), 3) uaktywnianie cyklazy guanylanowej i 4) rozszerzanie naczyń krwionośnych (20). Obecnie sądzi się, że tlenek azotu może spełniać tą samą funkcję co EDRF. Wiele badań dowodzi, że końcowym czynnikiem odpowiedzialnym za rozszerzanie naczyń krwionośnych, będącym wynikiem stosowania np: nitroprusydku sodu, czy nitrogliceryny, jest właśnie tlenek azotu (20).
Co ważniejsze, tlenek azotu może również powstawać endogennie. Jego prekursorem staje się arginina w cyklu mocznikowym. Katalizowana przez syntetazę tlenku azotu arginina jest najpierw hydroksylowana a następnie utleniana, w wyniku czego powstaje cytrulina i tlenek azotu. Koprodukt tej reakcji cytrulina jest później metabolizowana powtórnie do argininy tworząc pół cyklu mocznikowego (ryc. 1) (13, 24). W badaniach nad syntetazą tlenku azotu używano, jako jej antagonistów, strukturalnych analogów argininy np: NG – monometyl – L – argininę, ester metylowy NG – nitro – L – argininy i L – NG – nitro – argininę (16).
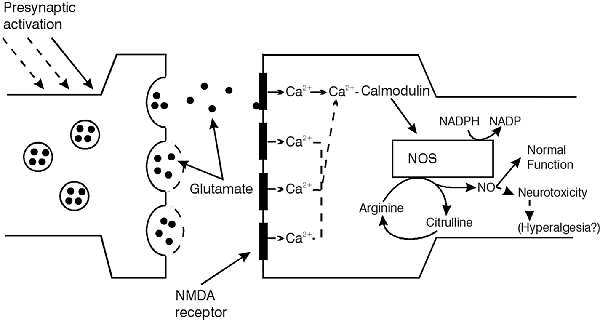
Rys. 1. Diagram ilustrujący przebieg reakcji aktywowanych przez syntezę tlenku azotu i sposobu powstawania tlenku azotu (NO w synapsie glutametergicznej (24).
Syntetaza tlenku azotu występująca w komórkach śródbłonka naczyniowego, makrofagach, neuronach różni się między sobą budową, tworząc izoformy. Wyizolowano cztery izoformy syntetazy tlenku azotu (11). Gen odpowiedzialny za ich wytwarzanie znaleziono w klonowanych neuronach mózgowia (11). Badając syntetazę tlenku azotu można pośrednio śledzić działanie tlenku azotu. Tak więc, skoro tlenek azotu jest tak trudny do badania (jest utleniany do azotynu i azotanu w kilka sekund po powstaniu) o wiele łatwiej jest badać enzym niż powstający tlenek azotu (16).
W centralnym układzie nerwowym, syntetazę tlenku azotu lokalizowano w neuronach na różnych jego piętrach włączając korę mózgu, hipokamp, móżdżek, rdzeń kręgowy. Najwyższy poziom m-RNA odpowiedzialnego za powstawanie syntetezy tlenku azotu znaleziono w móżdżku (8, 16). Zakłada się, że syntetaza tlenku azotu występująca w neuronach wymaga kalmoduliny jako kofaktora oraz zredukowanego fosforanu dwunukleotydu nikotynadeninowego (NADPH) jako dawcy elektronu.
Ostatnio aktywność syntetazy tlenku azotu została powiązana z receptorem kwasu N – metylo – D – asparginowego (NMDA) będącego podtypem aminokwasowego receptora pobudzającego. Syntetaza tlenku azotu jest wysoko zależna od stężenia jonów wapnia w neuronach. Stwierdzono brak katalitycznej aktywności, gdy wewnątrzkomórkowe stężenie jonów wapnia jest na poziomie spoczynkowym. Jak przedstawia rycina 1 aktywacja presynaptycznego glutametergicznego zakończenia zapoczątkowywuje uwalnianie pobudzającego neuroprzekaźnika glutaminianu. W następstwie wiązania glutaminianu z postsynaptycznymi receptorami NMDA powstaje pobudzenie receptora i napływ jonów wapnia do postsynaptycznego neuronu. Zostaje uformowany kompleks Ca++ – kalmodulina, syntetaza tlenku azotu jest aktywowana i syntetyzowany tlenek azotu (1, 6, 8, 16, 24).
Tlenek azotu nie jest klasycznym neuroprzekaźnikiem, ponieważ nie jest uwalniany bezpośrednio przez depolaryzację. Podstawowy kierunek działania tlenku azotu to podniesienie poziomu c-GMP (cyklicznego guanozyno 3´, 5´ – monofosforanu) w komórkach docelowych poprzez uaktywnienie cyklazy guanylanowej. Obwodowo tlenek azotu może działać jako neuroprzekaźnik, będąc wytwarzanym i uwalnianym presynaptycznie w zakończeniach nerwowych i dyfunując w ich otoczenie. Najlepszą fizjologiczną dokumentacją takiego działania tej synaptycznej aktywności jest relaksacja mięśni gładkich (np: w przewodzie pokarmowym, penisie) (16, 24).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Bennett G.J.: Chronic pain due to peripheral nerve damage: an overview. Progr. in Pain Res. and Management, Vol. 1, IASP Press, Seattle, 1994, s. 51-57. 2. Bredt D., Snyder S.: Nitric oxide, a novel neuronal messenger. Neuron, 1992, 8, s. 3-11. 3. Choi D.: Ketamine reduces NMDA receptor mediated neurotoxicity in cortical cultures. Status of Ketamine in Anesthesiology, 1990, NPP Books edited by E. F. Domino, s. 549-555. 4. Davar G. et al.: MK-801 blocks the development of thermal hyperalgesia in a rat model of experimental painful neuropathy. Brain Res., 1991, 553, s. 327-330. 5. Dawson V. et al.: Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc. Natl. Acad. Sci. USA, 1991, 88, s. 6358-6371. 6. Dickenson H.A.: NMDA receptor antagonists as analgesic. Progr. in Pain Res. and Management, Vol. 1, IASP Press, Seattle, 1994, s. 173-184. 7. Dickenson A., Sullivan A.: NMDA receptors and central hyperanalgesic states. Pain, 1991, 46, s. 344-345. 8. Dubner R.: Neuronal plasticity and pain following peripheral tissue inflammation or nerve injury. Proceeding of the VIth World Congress on Pain, Elsevier, Amsterdam, 1991, s. 263-276. 9. Dubner R., Ruda M.: Activity dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci., 1992, 15, s. 96-103. 10. Edelman G., Gally J.: Nitric oxide linking space and time in the brain. Proc. Natl. Acad. Sci. USA, 1992, 89, s. 11651-11652. 11. Forstermann U. et al.: Isoforms of nitric oxide synthase. Biochem. Pharmacol., 1991, 42, s. 1849-1859. 12. Gally J. et al.: The NO hypothesis: possible effects of a short-lived, rapidly diffusible signal in the development and function of the nervous system. Proc. Natl. Acad. Sci. USA, 1990, 87, s 3547-3551. 13. Garthwaite J.: Glutamate, nitric oxide and cell-cell signaling in the nervous system. TINS, 1991, 14, s. 60-67. 14. Gracely R. et al.: Painful neuropathy: altered central processing, maintained dynamically by peripheral input. Pain, 1992, 51, s. 175-194. 15. Haley J. et al.: Evidence for spinal N-methyl-D-aspartate receptor involvement in prolonged chemical nociception in the rat. Brain Res., 1990, 518, s. 218-226. 16. Kimes A. et al.: Nitroarginine, a nitric oxide synthase inhibitor, attenuates morphine withdrawal. Soc. Neurosci. Abs., 1991,17, s. 538. 17. Laird J. et al.: Dorsal root potentials and afferent input to the spinal cord in rats with an experimental peripheral neuropathy. Brain Res., 1992, 548, s. 181-190. 18. Mao J. et al.: Intrathecal treatment with dextrorphan or ketamine potently reduces pain – related behaviours in rat model of peripheral mononeuropathy. Brain Res., 1992, 576, s. 254-262. 19. Mc Quay H., Dickenson A.: Implications of central nervous system plasticity for pain management. Anaesthesia, 1990, 45, s. 101-102. 20. Meller S. et al.: Is there a role for an endothelium- derived relaxing factor in nociception? Brain Res., 1990, 531, s. 342-345. 21. Meller S., Gebhard G.: Nitric oxide (NO) and nociceptive processing in the spinal cord. Pain, 1993, 52, s. 127-136. 22. Raigorodsky G., Urca A.: Intrathecal N-methyl-d-aspartate (NMDA) activates both nociceptive and antinociceptive system. Brain Res., 1987, 422, s. 158-162. 23. Ren K. et al.: Effects of a noncompetitive NMDA receptor antagonist, MK-801, on behavioral hyperalgesia and dorsal horn neuronal activity in rats with unilateral inflammation. Pain, 1992. 24. Ren K. et al.: Nitric oxide: A role in neurotransmission or neuronal toxicity? IASP Newletter, 1992, March-April, s. 2-3. 25. Seltzer Z. et al.: Modulation of neuropathic pain in rat by spinal distribution and NMDA receptor blockade of injury discharge. Pain, 1991 (a), 46, s. 327-336. 26. Stanfa L. et al.: Increased potency of spinal opiates after peripheral inflammation in the halothane anaesthetized rat. Pain, 1992, 50, s. 345-354. 27. Woolf C., Thompson S.: The induction and maintenance of central sensitization is dependent on N-methyl-D-aspartic acid receptor activation; implications for the treatment of post-injury hypersensitivity states. Pain, 1991, 44, s. 293-299. 28. Yaksh T.: Behavioural and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor system and excitatory amino acid antagonists. Pain, 1989, 37, s. 111-123.