© Borgis - Nowa Pediatria 4/2014, s. 122-127
Tomasz Floriańczyk, Agnieszka Tomik, Sylwia Łuszczyk, Małgorzata Gołąbek-Dylewska, *Bożena Werner
Ocena przebiegu klinicznego wad serca u dzieci z zespołem Noonan
Cardiac malformations in children with Noonan syndrome
Klinika Kardiologii Wieku Dziecięcego i Pediatrii Ogólnej, Warszawski Uniwersytet Medyczny
Kierownik Kliniki: prof. dr hab. n. med. Bożena Werner
Summary
Introduction. Noonan syndrome is autosomal dominant anomaly characterized by variable, multiple, congenital malformations.
Aim. The aim of the study was an evaluation of congenital cardiovascular defects in children and teenagers with Noonan syndrome.
Material and methods. Study group consisted of 20 children who were diagnosed with Noonan syndrome based on typical phenotype symptoms. In addition in 3 patients diagnosis was confirmed using genetic test. In all patients physical examination, ECG tracing, X-ray picture of the chest, echocardiographic examination and 24-hours ECG Holter monitoring were performed.
Results. Pulmonary valvular stenosis was a major cardiovascular anomaly in study group and was diagnosed in 16 patients. In 10 of them the atrial septal defect was a coexisting anomaly. The balloon pulmonary valvuloplasty was performed in 4 patients. Satisfactory result of valvuloplasty was found in one case in contrary to the remain 3 with dysplastic pulmonary valves and coexisting supravalvular stenosis. Hypertrophic cardiomyopathy with left ventricular outflow tract obstruction was diagnosed in 1 patient.
Conclusions. Children suspected of Noonan syndrome require pediatric cardiologist opinion and in the case of confirmed diagnosis of Noonan syndrome permanent cardiologic care is mandatory.

WSTĘP
Zespół Noonan jest uwarunkowanym genetycznie zespołem wad wrodzonych, występującym z częstością 1:1000-1:2500 żywo urodzonych dzieci, bez preferencji płci.
Chorobę w większości przypadków dziedziczy się w sposób autosomalnie dominujący, a defekt genetyczny zlokalizowany jest u 30-60% pacjentów na chromosomie 12 w regionie 12q24.1 i dotyczy enzymu kinazy tyrozynowej typu 11 (1, 2).
Zespół Noonan jest zespołem patologii wielonarządowych, obejmujących układ sercowo-naczyniowy, limfatyczny, kostno-szkieletowy, moczowo-płciowy, krwiotwórczy oraz ośrodkowy układ nerwowy (3-5). Do cech fenotypowych zespołu Noonan zaliczamy: typową dysmorfię twarzy obejmującą hiperteloryzm, antymongoidalne ustawienie szpar powiekowych, opadanie powiek, nisko osadzone małżowiny uszne, szeroką podstawę nosa oraz krótką i płetwiastą szyję, ponadto niskorosłość, różnego stopnia upośledzenie umysłowe, wady wrodzone układu sercowo-naczyniowego, deformacje klatki piersiowej oraz wady postawy, wady wrodzone nerek oraz zaburzenia pokwitania.
CEL PRACY
Celem pracy była analiza patologii układu sercowo-naczyniowego u dzieci i młodzieży z zespołem Noonan, ze szczególnym uwzględnieniem objawów klinicznych i leczenia.
MATERIAŁ I METODY
Grupę badaną stanowiło 20 dzieci w wieku od 3 miesięcy do 15 lat, skierowanych do Kliniki Kardiologii z podejrzeniem wrodzonej patologii układu sercowo-naczyniowego.
Rozpoznanie zespołu Noonan u dzieci w badanej grupie ustalono na podstawie typowych cech fenotypowych oraz współistniejących patologii układu krążenia. Badaniem genetycznym potwierdzono rozpoznanie zespołu Noonan u 3 dzieci, w tym u 2 stwierdzono mutację genu PTPN1, a u jednego RAF1. U pozostałych 17 dzieci nie wykonywano badań genetycznych.
U wszystkich dzieci przeprowadzono badanie przedmiotowe oraz wykonano 12-odprowadzeniowe badanie EKG, badanie radiologiczne klatki piersiowej, badanie echokardiograficzne i całodobowy zapis EKG metodą Holtera.
WYNIKI
Wśród badanych było 11 chłopców i 9 dziewczynek. W badaniu przedmiotowym u wszystkich dzieci stwierdzono typowe dla zespołu Noonan cechy dysmorfii, ponadto u 15 pacjentów niski wzrost, u 9 kurzą klatkę piersiową, u 8 chłopców brak jąder w worku mosznowym i u jednego skrzywienie kręgosłupa. Analizując odchylenia od normy, w badaniu fizykalnym dominującym objawem był szmer nad sercem, który stwierdzono u wszystkich 20 dzieci, w tym u 15 u podstawy serca i u 5 wzdłuż lewego brzegu mostka.
U trojga dzieci, których matki były obarczone zespołem Noonan (objawy obecne również u babci jednego z dzieci) rozpoznano zespół rodzinny. Chorobami towarzyszącymi u jednego dziecka była niedoczynność tarczycy, u jednego stwierdzono zaburzenia krzepnięcia z niedoborem czynnika X, u 3 dzieci wykryto zdwojenie układu kielichowo-miedniczkowego nerek w badaniu USG jamy brzusznej.
W badaniu EKG u 12 dzieci nie stwierdzono odchyleń od normy. Spośród pozostałych pacjentów u 8 obserwowano cechy przerostu prawego przedsionka i prawej komory. U 9 oś elektryczna serca była odchylona w prawo, u 4 w lewo, a u pozostałych określana jako oś pośrednia (ryc. 1A, 2A). U 6 dzieci stwierdzono nieprawidłowy stosunek załamków R/S z dominacją załamków S w zespole QRS w odprowadzeniach przezklatkowych znad lewej komory serca (ryc. 1B, 2B). U jednego z dzieci odstęp QT i QTc był wydłużony (ryc. 1A, 1B). U jednej pacjentki zarejestrowano pojedyncze pobudzenia przedwczesne nadkomorowe.
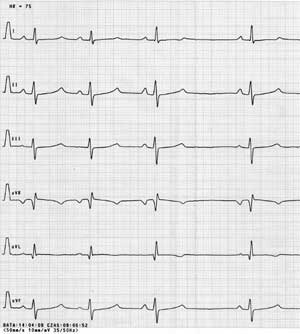
Ryc. 1A. EKG dziecka z kardiomiopatią przerostową – lewogram, wydłużenie odstępu QT.
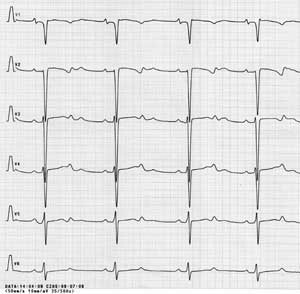
Ryc. 1B. EKG dziecka z kardiomiopatią przerostową – nieprawidłowy stosunek R/S w odprowadzeniach V5-V6, wydłużenie odstępu QT.
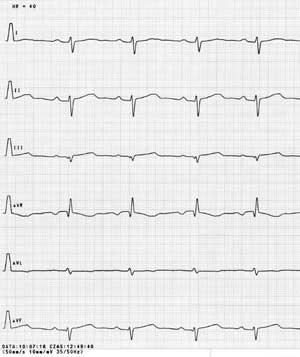
Ryc. 2A. EKG dziecka ze zwężeniem zastawki pnia płucnego – lewogram.
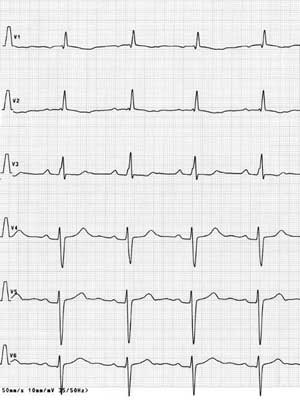
Ryc. 2B. EKG dziecka ze zwężeniem zastawki pnia płucnego – nieprawidłowy stosunek R/S w odprowadzeniach V5 i V6.
W badaniu holterowskim u 17 pacjentów nie stwierdzono zaburzeń rytmu serca, u 2 dzieci z zastawkowym zwężeniem pnia płucnego rejestrowano nieliczne (poniżej 1000/dobę) zaburzenia rytmu serca pod postacią pojedynczych pobudzeń przedwczesnych nadkomorowych i komorowych, u jednego dziecka z kardiomiopatią przerostową okresowo rejestrowano wolny rytm zatokowy i pojedyncze przedwczesne pobudzenia komorowe.
W badaniu radiologicznym klatki piersiowej u 11 dzieci wielkość sylwetki serca mieściła się w normie. U 9 stwierdzono powiększenie sylwetki serca w zakresie prawej komory serca. Rysunek naczyniowy płuc u wszystkich był prawidłowy.
Badaniem echokardiograficznym u 16 dzieci stwierdzono zwężenie zastawki pnia płucnego, z maksymalnym gradientem ciśnienia skurczowego pomiędzy prawą komorą a pniem płucnym wynoszącym 25-80 mmHg, średnio 41,6 mmHg (ryc. 3). Dodatkowo, u 10 dzieci ze zwężeniem zastawki pnia płucnego stwierdzono ubytek przegrody międzyprzedsionkowej, w tym u 3 dzieci ubytki mnogie (ryc. 4). U 3 z nich stwierdzono wiotkie płatki zastawki dwudzielnej bez niedomykalności. U dwojga dzieci wykryto izolowane ubytki przegrody międzyprzedsionkowej, u jednego częściową postać ubytku przegrody przedsionkowo-komorowej. U jednego dziecka rozpoznano kardiomiopatię przerostową ze zwężeniem drogi odpływu lewej komory serca z maksymalnym gradientem skurczowym 50 mmHg (ryc. 5A, 5B).
Ryc. 3. Badanie echokardiograficzne dziecka ze zwężeniem zastawki pnia płucnego (PS) z postenotycznym poszerzeniem (PS – zwężenie zastawki pnia płucnego; MPA – pień tętnicy płucnej; RPA – prawa gałąź tętnicy płucnej; LPA – lewa gałąź tętnicy płucnej).
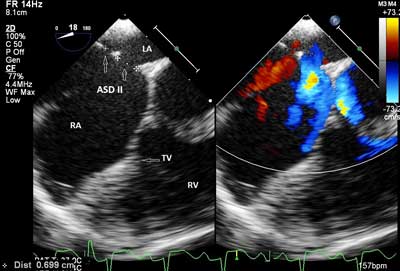
Ryc. 4. Przezprzełykowe badanie echokardiograficzne dziecka z mnogimi ubytkami przegrody międzyprzedsionkowej typu II (ASD II – ubytek przegrody międzyprzedsionkowej; RA – prawy przedsionek; LA – lewy przedsionek; RV – prawa komora; TV – zastawka trójdzielna).
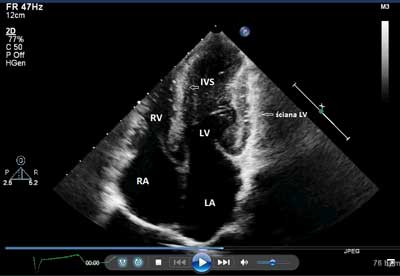
Ryc. 5A. Badanie echokardiograficzne dziecka z kardiomiopatią przerostową z widocznym pogrubieniem przegrody i wolnej ściany lewej komory serca (LV – lewa komora; RV – prawa komora; IVS – przegroda międzykomorowa; LA – lewy przedsionek; RA – prawy przedsionek).
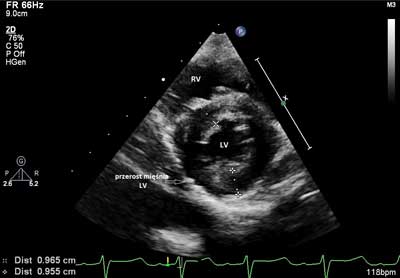
Ryc. 5B. Badanie echokardiograficzne dziecka z kardiomiopatią przerostową z koncentrycznym przerostem mięśnia lewej komory (LV – lewa komora; RV – prawa komora).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Mendez H, Opitz J: Noonan syndrome: a review. Am J Med Genet 1985; 21: 493-506. 2. Tartaglia M, Pennacchio L, Zhao C et al.: Gain-of-function SOS1 mutation cause a distinctive form of Noonan syndrome. Nat Genet 2007; 39: 75-79. 3. Krajewska-Walasek M: Zespół Noonan w praktyce klinicznej. Ped Pol 1995; 70(10): 809-817. 4. Gawlik A, Koehler B, Wackerman-Ramos A, Gawlik T: Kryteria diagnostyczne dla zespołu Noonan. Ped Pol 2000; 75: 419-423. 5. Bhambhani V, Muenke M: Noonan syndrome. Am Fam Physician 2014; 89: 7-43. 6. Allanson J: Noonan Syndrome. Am J Med Genet Part C Semin Med Genet 2007; 145C: 274-279. 7. Jorge A, Malquias A, Arnhold I, Menonca B: Noonan Syndrome and related Disorders: a review of Clinical Features and Mutations in genes of the RAS/MAPK Pathway. Horm Res 2009; 71: 185-193. 8. Colquitt J, Noonan J: Cardiac Findings in Noonan Syndrome on Long-term Follow-up. Congenit Heart Dis 2014; 9: 144-150. 9. Romano A, Allanson J, Dahlgren J et al.: Noonan Syndrome: Clinical Features, Diagnosis, and Management Guidelines. Pediatrics 2010; 126: 746-760. 10. Kucińska B, Werner B, Godlewski K et al.: Problemy kardiologiczne u dziecka z zespołem Noonan. Standardy Medyczne Pediatria 2009; 83-88. 11. Werner B, Penconek K, Wróblewska-Kałużewska M: Wrodzone wady serca u dzieci z zespołem Noonan. Ped Pol 2002; 77: 683-687. 12. Bochyńska A, Ziółkowska L: Kardiomiopatia przerostowa u noworodka z rzadkim zespołem genetycznym – trudności diagnostyczne i terapeutyczne. Postępy Nauk Medycznych 2014; 27: 652-657. 13. Raaijmakers R, Noordam C, Noonan JA et al.: Are ECG abnormalities in Noonan syndrome characteristic for the syndrome? Eur J Pediatr 2008; 167: 1368-1367. 14. Ucar T, Atala S, Tekin M, Tutar E: Bilateral Coronary Artery Dilatation and Supravalvular Pulmonary Stenosis in a Child with Noonan Syndrome. Pediatr Cardiol 2005; 26: 848-850. 15. Cornwall J, Green R, Nielsen J, Gelb B: Frequency of Aortic Dilation in Noonan Syndrome Am J Cardiol 2014; 113: 368-371. 16. Noordam C: Growth Hormone and the Heart in Noonan Syndrome. Horm Res 2009; 72 (suppl. 2): 49-51. 17. Ranke M: Noonan Syndrome: Growth to Growth Hormone – The Experience of Observational Studies. Horm Res 2009; 72 (suppl. 2): 36-40.