© Borgis - Postępy Nauk Medycznych 8/2015, s. 622-626
*Renata Posmyk1, Beata Bugała-Musiatowicz2, Ryszard Leśniewicz1, Magdalena Gogiel1, Izabela Radowska3, Marcin Musiatowicz4, Izabela J. Szarmach2
Anomalie zębowe oraz obraz kliniczny 10-letniej dziewczynki z częściową trisomią 22q13->qter
Orodental anomalies and clinical description in 10 years-old girl with a partial trisomy 22q13->qter
1Department of Clinical Genetics, Podlaskie Center of Clinical Genetics, Białystok
Head of Department: Ryszard Leśniewicz, MD, PhD
2Department of Orthodontic, Medical University, Białystok
Head of Department: Izabela Szarmach, MD, PhD
3Department of Pediatric Ophthalmology and Strabismus, Children’s University Hospital, Białystok
Head of Department: prof. Alina Bakunowicz-Łazarczyk, MD, PhD
4Department of Pediatric Otorhinolaryngology, Medical University, Białystok
Head of Department: prof. Elżbieta Hassmann-Poznańska, MD, PhD
Streszczenie
Głównym celem pracy jest prezentacja anomalii zębowych oraz obrazu klinicznego u 10-letniej pacjentki z częściową trisomią 22q13->qter. Do głównych objawów stwierdzonych u dziecka zaliczyliśmy: znacznie opóźniony rozwój psychoruchowy, niski wzrost, słabe przybieranie na masie, wadę serca, obustronne przepukliny pachwinowe oraz zespół wybitnych cech dysmorfii twarzy (małogłowie, retrognatyzm, długi odstęp nosowo-wargowy, małe, nisko osadzone i do tyłu zrotowane uszy, skośnie w dół skierowane szpary powiekowe, zmarszczki nakątne, ptoza powiek oraz szeroka szpara ust).
Badanie wewnątrzustne wykazało wadę zgryzu klasy III, wąskie wysokie podniebienie z rozszczepem podniebienia miękkiego, wąski dolny łuk zębowy, opóźnione wyżynanie zębów stałych oraz anomalie dotyczące kształtu zębów. Zdjęcie rtg pantomograficzne wykazało wrodzony brak zawiązków zębów stałych, w tym pierwszego zęba trzonowego po stronie prawej, bocznego siekacza prawego w obrębie szczęki oraz bocznego siekacza żuchwy również po stronie prawej.
Istnieją jedynie pojedyncze doniesienia o anomaliach zębowych łącznie ze szczegółowym opisem cech dysmorfologicznych u dzieci z rzadkimi zespołami chromosomowymi. Szczególnie rzadko opisywane są częściowe trisomie. Tłumaczyć to można tym, że zazwyczaj do szerokiego spektrum cech klinicznych należą ciężkie wady wrodzone, które znacznie skracają przeżycie osób z tą aberracją chromosomową. Zwracamy uwagę na potrzebę kolejnych doniesień dotyczących anomalii zębowych w trisomii 22, które są bardzo rzadko opisywanym zjawiskiem.
Summary
We report orodental manifestations and clinical description in a patient with a pure partial trisomy 22q13->qter. The 10 years-old girl was evaluated by the same physicians from the birth up to date. The main characteristic features of the patients consisted of severe psychomotor retardation, short stature, failure to thrive, heart defect, bilateral inguinal hernias and a set of peculiar dysmorphic features (microcephaly, maxillary retrognathism, long philtrum, small, low set and posteriorly rotated ears, down slanting palpebral fissures, epicanthal folds, ptosis and wide mouth fissure). The intraoral examination revealed class III malocclusion, narrow high-arched palate with clefting, narrow lower dental arch, delayed eruption of permanent dentition and teeth shape abnormalities. Panoramic radiograph showed congenitally missing permanent maxillary first molar on right side, maxillary lateral incisors and lateral incisor in mandible on right side.
Reports of dental anomalies along with detail dysmorphic features description in children with rare chromosome syndromes caused by autosomal trisomies are unique findings. Usually a clinical spectrum of such abnormalities consists of a wide range of other severe congenital malformations leading towards significantly reduced life expectancy. We stress the need of further detail presentation of dental problems in trisomy 22, because it is still a very rare event in medical publications.

Introduction
The genomic disorders associated with chromosome 22 are clinically quite well defined, but dental descriptions remain a rare finding. Trisomy of chromosome 22 belongs to a group of rare chromosomal aberrations in newborns. It was first well documented and described in 1971 by Hsu et al. (1). Trisomy 22 may exist in mosaic, complete non-mosaic or in partial forms. Complete trisomy 22 was seen commonly in spontaneous abortuses (2, 3). Among live births it has been rarely reported (4-7). The mosaic forms were described in several publications (8-10). It was observed, that mosaic trisomy 22 are compatible with better prognosis for survival, while complete non-mosaic are rather lethal. Dysmorphic features and clinical signs of patients with complete and mosaic trisomies were described in a several publications (10, 11). Phenotypic descriptions of partial trisomies are uncommon and usually they are very variable due to influence of other chromosome. Pure partial trisomies of chromosome 22 are rather rare findings (12, 13).
We present a 10 years follow-up of a unique patient with the pure partial trisomy 22q13->qter. Detail phenotype descriptions with emphasis on dental problems are of increased value. Dental abnormalities due to different chromosomal trisomies are rarely described because of a poor lifespan prognosis. It also requires a good cooperation between patient’s family and many medical care providers (clinical geneticists, pediatricians, dentists, orthodonticians, cardiologists, etc.). We have not found any follow-up report regarding orodental problems in a child with trisomy 22.
In a child with genetic disorder is especially difficult to set up appropriate management of dental problems. Usually dental abnormalities come along with other birth defects (e.g. heart, gastrointestinal or CNS problems) and a variable degree of mental retardation. An appropriate treatment planning is essential in accuracy of diagnosis. Craniofacial growth charts are different among many genetic syndromes. Parental support and understanding of harmful because of intensity, frequency, and duration of treatment process is of great value. It is not possible in rare genetic disorders, where phenotypic spectrum is not well defined.
Case report
A female patient was admitted to genetic counseling unit at the age of one month due to a set of distinct dysmorphic features, hypotonia and other major congenital malformations. A girl was born from the second pregnancy, by normal vaginal delivery at the 37th week of pregnancy to young, healthy and non-consanguineous parents. IUGR (Intrauterine Growth Retardation) and polyhydramnions were detected in prenatal ultrasound examination at 28 weeks of gestation. Birth biometry was: weight 2440 g, length 47 cm, head circumference 32 cm, thorax circumference 30 cm and Apgar score 6. Cleft hard and soft palate, pes equinovarus, hypotonia and unusual face were described during first examination by a neonatologist. Respiratory distress syndrome was noticed shortly after birth. Hypoglycemia, hypocalcaemia, severe bilirubinemia and ASD III° were diagnosed at the 3rd day of life. Initially poor lifespan prognosis was given, but the girl was getting better from day to day. Finally she spent 10 days at the hospital. Intensive rehabilitation was used from the first days. She was operated for bilateral inguinal hernia and palate clefting during first 12 months of life. Gastroesophageal reflux was treated. Orthopedic intervention for pes equinovarus was necessary. Ophthalmological examination did not revealed typical cat eye sign. Strabismus and hyperopia were detected. She was operated for strabismus at the age of two years. Recurrent bilateral otitis media were noticed. Tympanometry revealed type B curve in right and left ear.
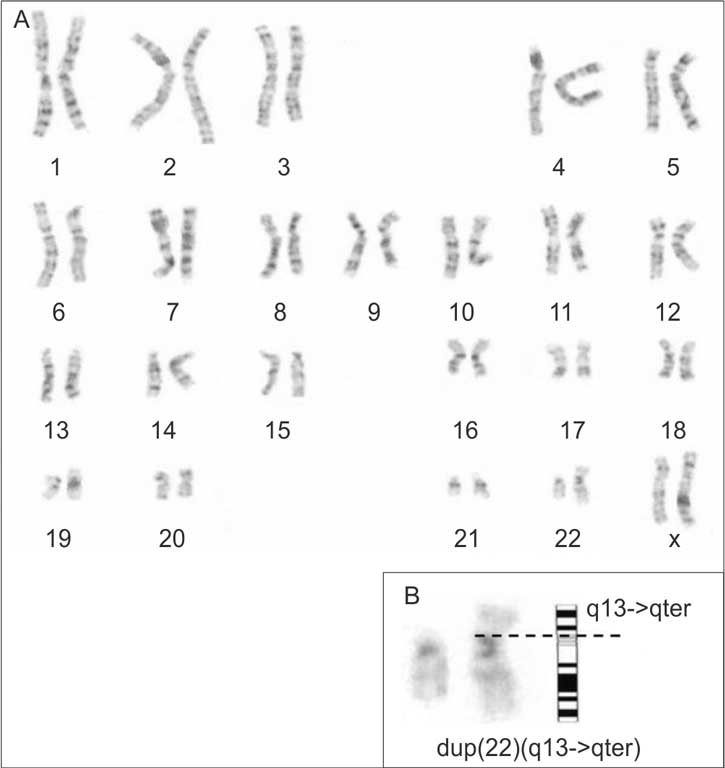
Genetic testing was suggested by clinical geneticist. Karyotype from lymphocytes was initially established as 46,XX, add(22)(p11) using standard GTG banding technique. Large satellites at the short arms (p) of chromosome 22 were detected. It was excluded by CTG banding. M-FISH (Multiplex Fluorescence in Situ Hybridization) testing confirmed that extra material on chromosome 22 comes from the same chromosome 22, because showed the chromosome 22 specific spectral signature. In further FISH analysis with 22q specific probes was evaluated that additional material was derivered from the long arm of chromosome 22. Final karyotype was: 46,XX,dup(22)(q13->qter) (fig. 1). Karyotypes of both parents were normal.
Fig. 1. (A) Karyotype and (B) partial karyotype of dup(22)(q13->qter).
Her psychomotor development was delayed. She began to walk at 2 years, to say a few simple words and sentences at 4 years. Her psychological test at 6 years estimated her nonverbal intelligence as II = 57 (by Leiter P-93 scale), social development was below average IDS = 77 (by Doll scale). Her expressive language was retarded according AFA-scale. She underwent cardio surgical operation for ASD at the age of 10 years.
Orthodontic care management was started from the age of 4 years.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Hsu LY, Shapiro LR, Gertner M et al.: Trisomy 22: a clinical entity. J Pediatr 1971; 79: 12-19.
2. Hassold T, Chen N, Funkhouser J et al.: A cytogenetic study of 1000 spontaneous abortions. Ann Hum Genet 1980; 44: 151-178.
3. Bacino CA, Schreck R, Fischel-Ghodsian N et al.: Clinical and molecular studies in full trisomy 22: further delineation of the phenotype and review of the literature. Am J Med Genet 1995; 56: 359-365.
4. McPherson E, Stetka DG: Trisomy 22 in a liveborn infant with multiple congenital anomalies. Am J Med Genet 1990; 36: 11-14.
5. Feret MA, Galán F, Aguilar MS et al.: Full trisomy 22 in a malformed newborn female. Ann Genet 1991; 34: 44-46.
6. Kobrynski L, Chitayat D, Zahed L et al.: 22 and facioauriculovertebral (Goldenhar) sequence. Am J Med Genet 1993; 46: 68-71.
7. Mihçi E, Taçoy S, Yakut S et al.: Maternal origin and clinical findings in a case with trisomy 22.Turk J Pediatr 2007; 49: 322-326.
8. Wertelecki W, Breg WR, Graham JM Jr et al.: Trisomy 22 mosaicism syndrome and Ullrich-Turner stigmata. Am J Med Genet 1986; 23: 739-749.
9. Woods CG, Bankier A, Curry J et al.: Asymmetry and skin pigmentary anomalies in chromosome mosaicism. J Med Genet 1994; 31: 694-701.
10. Crowe CA, Schwartz S, Black CJ, Jaswaney V: Mosaic trisomy 22: a case presentation and literature review of trisomy 22 phenotypes. Am J Med Genet 1997; 71: 406-413.
11. Basaran N, Berkil H, Ay N et al.: A rare case: mosaic trisomy 22. Ann Genet 2001; 44: 183-186.
12. Prasher VP, Roberts E, Norman A et al.: Partial trisom22 (q11.2-q13.1) as a result of duplication and pericentric inversion. J Med Genet 1995; 32: 306-308.
13. Wieczorek D, Holtvogt J, Thonig S, Gillessen-Kaesbach G: A female patient with partial duplication 22 (q13->qter). Clin Dysmorphol 1998; 7: 289-294.
14. McNamara JA Jr: A method of cephalometric evaluation. Am J Orthod 1984; 86: 449-469.
15. Fujimoto A, Wilson MG, Towner JW: Duplication of the segment q12.2 leads to qter of chromosome 22 due to paternal inversion 22(p13q12.2). Hum Genet 1983; 63: 82-84.
16. Rivera H, Garcia-Esquivel L, Romo MG et al.: The 22q distal trisomy syndrome in a recombinant child. Ann Genet 1988; 31(1): 47-49.
17. Abeliovich D, Maor E, Bashan N, Carmi R: Duplication of distal 22q. Am J Med Genet 1989; 32: 346-349.
18. Mirza G, Imaizumi K, Ragoussis J: Partial trisomy 22 in a liveborn resulting from a rearrangement between chromosomes 6 and 22. J Med Genet 2000; 3: E22.
19. Slater HR, Voullaire LE, Vaux CE et al.: Confirmation of trisomy 22 in two cases using chromosome painting: comparison with t(11;22). Am J Med Genet 1993; 46: 434-437.
20. Berends MJ, Tan-Sindhunata G, Leegte B, van Essen AJ: Phenotypic variability of Cat-Eye syndrome. Genet Couns 2001; 12: 23-34.