© Borgis - Postępy Nauk Medycznych 10/2015, s. 734-737
*Ryszard Grenda
Autosomalna dominująca wielotorbielowatość nerek (ADPKD) – cele farmakoterapii i powikłania pozanerkowe
Autosomal dominant polycystic kidney disease (ADPKD) – targets of pharmacotherapy and extrarenal complications
Department of Nephrology, Kidney Transplantation and Hypertension, The Children’s Memorial Health Institute, Warszawa
Head of Department: prof. Ryszard Grenda, MD, PhD
Streszczenie
Autosomalna dominująca wielotorbielowatość nerek jest jednym z wrodzonych schorzeń przebiegających z tworzeniem się torbieli narządowych, wykazującym głównie nerkową manifestację kliniczną (powiększenie objętości nerek, bóle okolicy lędźwiowej, krwiomocz, nadciśnienie tętnicze i niewydolność nerek). W niektórych przypadkach występują także objawy pozanerkowe, w postaci obecności torbieli wątroby, tętniaków naczyń mózgowych i aorty oraz uchyłków jelita grubego. Tło genetyczne jest skutkiem mutacji genów PKD1 i PKD2, które kodują swoiste produkty białkowe, znane jako policystyna 1 i 2. Niewydolność nerek przede wszystkim rozwija się w 6. dekadzie życia. Niezależnie od tradycyjnego postępowania zachowawczego stosowanego w przewlekłej chorobie nerek, podejmowane są próby stosowania (w ramach badań klinicznych) celowanej terapii ukierunkowanej na wybrane elementy patomechanizmu tej choroby, zawierające wykorzystywanie inhibitorów konwertazy angiotensyny (ACE), blokerów szlaku mTOR, receptorów dla wazopresyny (V2) oraz inhibitorów cAMP. Żaden z tych sposobów leczenia nie okazał się jednoznacznie skuteczny w zakresie zahamowania powstawania i powiększania się torbieli jednocześnie z powstrzymaniem postępu niewydolności nerek bądź też częstość i nasilenie objawów niepożądanych były zbyt duże. Nadal konieczne jest prowadzenie kolejnych badań, mających na celu stworzenie skutecznej, ale jednocześnie akceptowalnej klinicznie terapii autosomalnej dominującej wielotorbielowatości nerek.
Summary
Autosomal dominant polycystic kidney disease (ADPKD) is hereditary cystic disorder with predominantly renal manifestation (large kidneys, flank pain, haematuria, hypertension and renal insufficiency). Some patients also present extrarenal symptoms, including hepatic cysts, cerebral and aortic aneurysms and colonic diverticula. The underlying genetic background of ADPKD is related to mutation of the PKD1 and PKD2 genes, coding specific protein products, known as polycystin 1 and 2. Renal failure develops mainly about the 6th decade of life. Apart from classic, symptomatic treatment of chronic kidney disease, several specific therapies, aimed to several pathways of disease mechanism, have been conducted in clinical trials, including use of ACE, mTOR, V2 receptor and cAMP inhibitors. None of those was universally effective in terms of complete stopping cysts growth and slowing deterioration of renal function or appeared to be not widely acceptable, due to high incidence of specific adverse events. Further investigation is required for developing effective and acceptable and friendly specific therapies of ADPKD.

Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is hereditary cystic disorder with predominantly renal manifestation (large kidneys, flank pain, haematuria, hypertension and renal insufficiency). Overall incidence adult population is 1/500-1/1000. Renal cysts are localized in all nephron segments (1). Ultrasonography (USG) is a major screening diagnostic tool and Ravine criteria are very simple and effective in preliminary diagnosis, as presented in table 1 (2). Arterial hypertension may precede the development of chronic kidney disease, as independent factor, related to renal volume and number of renal cysts. There is variable incidence of extrarenal comorbidities, including vascular wall pathologies localized in brain or aorta, prolapse of mitral valve, development of hepatic cysts and colonic diverticula. The overall incidence of specific symptoms and comorbidities of ADPKD is presented in table 2 (3-5). These data clearly show, that the age of the patient and longer duration of the disease are crucial to develop relevant overt symptoms and they are present more frequently in adult than in pediatric cases. Similar time correlation is relevant to the development of chronic kidney disease and then end-stage renal failure. To some extend it depends on gene mutation, as in PKD1 the end-stage renal failure develops about one and a half of the decade earlier, than in PKD2 mutation; (mean 53 vs 69 years), which probably is related to the final number of developing cysts, however in general the renal function is stable within first 40-50 years of age (when the cysts develop and grow) and then, in the stage of inflammation and fibrosis of renal parenchyma, the chronic renal disease accelerates and patients loose the GFR beyond the age of 50 (1, 6-9). This is related to the final cysts growth and cumulated, final kidney volume (10). The dynamics of this process is presented in figure 1 (9).
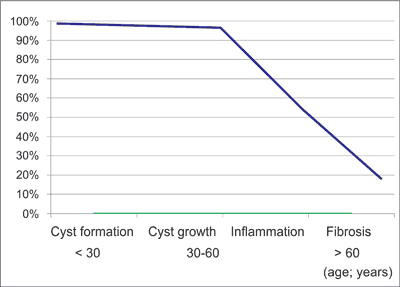
Fig. 1. Deterioration of GFR (% from baseline) in course of ADPKD.
Table 1. Ravine ultrasonographic criteria for screening the ADPKD (2).
| Age (years) | Positive family history | Negative family history |
| < 30 | 2 cysts bilaterally or unilaterally | 5 cysts bilaterally |
| 30-60 | 4 cysts bilaterally | 5 cysts bilaterally |
| > 60 | 8 cysts bilaterally | 8 cysts bilaterally |
Table 2. Clinical manifestation including extrarenal comorbidity of ADPKD in adults and children (3-5).
| Symptom | ADPKD in adults
(% of cases) | ADPKD in children
(% of cases) |
| Haematuria | 35-50 | 10 |
| Urine concentrating defect | 100 | 60 |
| Proteinuria | 18 | 14 |
| Nephrolithiasis | 20 | Not known |
| Flank/abdominal pain | 60 | 10 |
| Hepatic cysts | 83 | Up to 55 |
| Colonic diverticula | 82 | Not known |
| Cerebral aneurysms | 5-7 | < 5 |
| Prolapse of the mitral valve | 26 | 12 |
| Hypertension preceding loss of renal function | 60 | 22 |
Extrarenal symptoms in ADPKD patients
Liver cysts and intestinal diverticula
There are two major types of liver cysts – one, related to ADPKD and PKD1/PKD2 genes mutations, with prevalence of 0.2% and the second, rare isolated polycystic liver disease (prevalence < 0.01%), related to distinct (from ADPKD) gene mutation (gene PRKCSH; encoding hepatocystin). Similarly as in ADPKD, advancing age of the patient is a risk factor for liver – cyst growth and dysfunction (11). Colonic diverticula are quite common in adult ADPKD patients, however they are mainly asymptomatic. In cases of inflammation (diverticulitis) they become very severe clinical problem with high mortality rate (12). This pathology may be also localized in duodenum (13).
Vascular pathology in ADPKD
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Igarashi P, Somlo S: Genetics and pathogenesis of polycystic kidney disease. I Am Soc Nephrol 2002; 13: 2384-2398.
2. Braun WE: Autosomal dominant polycystic kidney disease: emerging concepts of pathogenesis and new treatments. Cleveland Clin J Med 2009; 76(2): 97-104.
3. Rizk D, Chapman A: Treatment of autosomal dominant polycystic kidney disease (ADPKD): the new horizon for children with ADPKD. Ped Nephrol 2008; 23(7): 1029-1036.
4. Fick-Brosnahan GM, Tran ZV, Johnson AM et al.: Progression of autosomal-dominant polycystic kidney disease in children. Kidney Int 2001; 59: 1654-1662.
5. Seeman T, Dusek J, Vondrichova H et al.: Ambulatory blood pressure correlates with renal volume and number of renal cysts in children with autosomal dominant polycystic kidney disease. Blood Pres Monit 2003; 8: 107-110.
6. Harris PC, Bae KT, Rosenti S et al.: Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2006; 17: 3013-3019.
7. Bae KT, Zhu F, Chapman AB et al.: Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease (CRISP): Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol 2006; 1(1): 64-69.
8. Hateboer N, van Dijk N, Bogdanova N et al.: Comparison of phenotypes of polycystic kidney diseases types 1 and 2. European PKD1-PKD2 Study Group. Lancet 1999; 353: 103-107.
9. Grantham JJ: Rationale for early treatment of polycystic kidney disease. Ped Nephrol 2015; 30: 1053-1062.
10. Fick-Brosnahan GM, Belz MM, McFann KK et al.: Relationship between renal volume growth and renal function in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2002; 39: 1127-1134.
11. Abu-Wassel B, Walsh C, Keough V, Molinari M: Pathopysiology, epidemiology, classification and treatment options for polycystic liver diseases. World J Gastroenterol 2013; 19(35): 5775-5786.
12. Pourfarziani V, Mousiavi-Nayeeni S-M, Ghaheri H et al.: The outcome of diverticulosis in kidney recipients with polycystic kidney disease. Transplant Proc 2007; 39: 1054-1056.
13. Kumar S, Adeva M, King BF et al.: Duodenal diverticulosis in autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 2006; 21: 3576-3578.
14. Rossetti S, Harris PC: The genetics of vascular complications in autosomal dominant polycystic kidney disease (ADPKD). Curr Hypertens Rev 2013; 9(1): 37-43.
15. Kim K, Drummond I, Ibraghimov-Beskrovnaya O et al.: Polycystin 1 is required for the structural integrity of blood vessels. PNAS 2000; 97(4): 1731-1736.
16. Neumann H, Malinoc A, Bacher J et al.: Characteristics of intracranial aneurysms in the Else-Kröner-Fresenius Registry for ADPKD. Cerebrovasc Dis Extra 2012; 2: 71-79.
17. Xu HW, Yu SQ, Mei CL et al.: Screening for intracranial aneurysm in 355 patients with ADPKD. Stroke 2011; 42: 204-206.
18. Hassane S, Claij N, Lantiga-van Leeuwen IS et al.: Pathogenic sequence for dissecting aneurysm formation in a hypomorphic polycystic kidney disease 1 mouse model. Arterioscler Thromb Vasc Biol 2007; 27: 2177-2183.
19. Torra R, Nicolau C, Badenas C et al.: Abdominal aortic aneurysms and autosomal dominant polycystic kidney disease. J Am Soc Nephrol 1996; 7: 2483-2486.
20. Paytner HE, Parnham A, Feest TG, Dudley CRK: Thoracic aortic dissection complicating autosomal dominant polycystic kidney disease. Nephrol Dial Transplant 1997; 12: 1711-1713.
21. Kim J, Kim SM, Lee SY et al.: A case of severe aortic valve regurgitation caused by an ascending aortic aneurysm in a young patient with autosomal dominant polycystic kidney disease and normal renal function. Korean Circ J 2012; 42: 136-139.
22. Patel MD, Ibarra PG, Rodriguez-Castro CE et al.: Multiple aortic aneurysms in a patient with adult polycystic kidney disease. Arch Salud Sin 2012; 6(4): 115-117.
23. Keuleers S, Verbeken E, Sinnaeve P: Aortic dissection with segmental mediolysis in polycystic kidney disease. Eur J Int Med 2009; 20: e9-e11.
24. Serra AL, Poster D, Kistler AD et al.: Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N Eng J Med 2010; 363: 820-829.
25. Waltz G, Budde K, Marwan I et al.: Everolimus in patients with in autosomal dominant polycystic kidney disease. N Eng J Med 2010; 363: 830-840.
26. Grantham JJ, Bennett WM, Perrone RD: mTOR inhibitors and autosomal dominant polycystic kidney disease. N Eng J Med 2011; 364(3): 286-287.
27. Torres VE, Chapman AB, Grantham JJ et al. and for the TEMPO Trial Investigators: Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Eng J Med 2012; 367(25): 2407-2418.
28. Erickson KF, Chertow GM, Goldhaber-Fiebert JD: Cost-effectiveness of tolvaptan in autosomal dominant polycystic kidney disease. Ann Intern Med 2013; 159(6): 382-389.
29. Caroli A, Perico N, Perna A et al. and for the ALADIN study group: Effect of longacting somatostatin analogue on kidney and cyst growth in autosomal dominant polycystic kidney disease (ALADIN): a randomised, placebo-controlled, multicentre trial. Lancet 2013; 382: 1485-1495.
30. Torres VE, Abebe KZ, Chapman A et al. and for the HALT-PD Trial Investigators: Angiotensin blockade in late polycystic kidney disease. N Eng J Med 2014; 371(24): 2267-2276.