© Borgis - Postępy Nauk Medycznych 1/2016, s. 22-30
*Monika Szturmowicz, Aneta Kacprzak
Rozpoznawanie i diagnostyka różnicowa nadciśnienia płucnego
Recognition and differential diagnosis of pulmonary hypertension
I Klinika Chorób Płuc, Instytut Gruźlicy i Chorób Płuc, Warszawa
Kierownik Kliniki: prof. dr hab. med. Jan Kuś
Streszczenie
Nadciśnienie płucne (ang. pulmonary hypertension – PH) jest stanem hemodynamicznym i patofizjologicznym, w którym średnie spoczynkowe ciśnienie w tętnicy płucnej mierzone w spoczynku, podczas cewnikowania prawego serca, osiąga lub przekracza wartość 25 mmHg. PH może rozwinąć się w przebiegu licznych chorób, rzadziej pojawia się bez uchwytnej przyczyny i wówczas określane jest jako idiopatyczne tętnicze nadciśnienie płucne. Niezależnie od etiologii, PH powoduje podobne, niespecyficzne objawy kliniczne, takie jak duszność i upośledzenie tolerancji wysiłku. Postępująca przebudowa naczyń płucnych prowadzi do wzrostu naczyniowego oporu płucnego, co skutkuje rozwojem niewydolności prawej komory serca. Rozpoznanie i prawidłowo przeprowadzona diagnostyka różnicowa PH mają kluczowe znaczenie dla dalszego postępowania leczniczego. Choć zalecane jest, żeby diagnostyka i leczenie odbywały się w ośrodkach wyspecjalizowanych w tej dziedzinie, to bardzo ważna jest znajomość tego zagadnienia zarówno przez lekarzy podstawowej opieki zdrowotnej, jak i lekarzy klinicystów różnych specjalizacji. W pracy prezentujemy aktualną wiedzę na temat rozpoznawania i diagnostyki różnicowej PH, zwracając uwagę na zmiany, jakich dokonano na V Światowym Kongresie Nadciśnienia Płucnego w 2013 roku oraz w wytycznych ERS/ESC zaprezentowanych we wrześniu 2015 roku.
Summary
Pulmonary hypertension (PH) is a hemodynamic and pathophysiological condition characterized by mean resting pulmonary artery pressure ≥ 25 mmHg, measured during right heart catheterization. PH may develop in the course of many diseases, less frequently it appears without any known underlying condition. On such occasion it is called idiopathic arterial pulmonary hypertension. Regardless of its origin, PH causes similar non-specific symptoms, such as dyspnoea and exercise intolerance. Progressive pulmonary vascular remodelling combined with growing pulmonary vascular resistance, lead to right heart failure. It is recommended that comprehensive diagnostic workup and treatment of PH should be performed at expert centre. Nevertheless, general practitioners and clinicians of various specialities should be familiar with this subject as well. Below we present current state of art concerning the recognition and differential diagnosis of pulmonary hypertension, highlighting changes that were made at The Fifth World Symposium on Pulmonary Hypertension in 2013 and the subsequent ERS/ESC guidelines presented in September 2015.

Definicja nadciśnienia płucnego
Nadciśnienie płucne (ang. pulmonary hypertension – PH) definiowane jest jako stan hemodynamiczny, w którym średnie ciśnienie w tętnicy płucnej mierzone w spoczynku podczas cewnikowania prawej części serca jest równe lub większe niż 25 mmHg (1).
Przyczynami nadciśnienia płucnego mogą być:
– patologia tętnic płucnych małego kalibru (tętnicze nadciśnienie płucne),
– upośledzona czynność lewej komory serca (żylne nadciśnienie płucne),
– choroby układu oddechowego (nadciśnienie płucne w przebiegu hipoksji i/lub zmian śródmiąższowych),
– zatorowość płucna (zatorowo-zakrzepowe nadciśnienie płucne),
– inne i złożone mechanizmy.
Podział nadciśnienia płucnego
Aktualny podział schorzeń przebiegających z nadciśnieniem płucnym, uwzględniający zmiany dokonane podczas V Międzynarodowego Kongresu Nadciśnienia Płucnego (Nicea 2013) (2), został przedstawiony na rycinie 1.

Ryc. 1. Nowa klasyfikacja nadciśnienia płucnego (Nicea 2013)
Grupa 1 dotyczy przypadków tętniczego nadciśnienia płucnego (ang. pulmonary arterial hypertension – PAH). Jest to nadciśnienie płucne przedwłośniczkowe (prekapilarne), przebiegające z prawidłowym ciśnieniem zaklinowania w tętnicy płucnej (≤ 15 mmHg) oraz z podwyższonym naczyniowym oporem płucnym (> 3 jednostek Wooda) (3). Według danych z rejestrów, 40-50% przypadków PAH stanowi idiopatyczne tętnicze nadciśnienie płucne (ang. idiopathic pulmonary arterial hypertension – IPAH), 20-30% – PAH w przebiegu chorób układowych tkanki łącznej, 11-23% – PAH w przebiegu wrodzonych/skorygowanych wad serca, 10% – PAH w przebiegu nadciśnienia wrotnego, 10% – PAH w przebiegu stosowania leków (4). Szczególnie niepokojące jest ciągłe poszerzanie się listy leków podejrzewanych o związek z nadciśnieniem płucnym. Doniesienia ostatnich lat udokumentowały wpływ dasatynibu, inhibitora kinazy tyrozynowej stosowanego w leczeniu przewlekłej białaczki szpikowej, na rozwój nadciśnienia płucnego u dorosłych oraz związek pomiędzy przetrwałym nadciśnieniem płucnym noworodków a stosowaniem przez matki inhibitorów wychwytu zwrotnego serotoniny w ciąży (2). Wykaz leków o udowodnionym lub prawdopodobnym wpływie na rozwój nadciśnienia płucnego przedstawiono w tabeli 1.
Tabela 1. Leki/toksyny związane z nadciśnieniem płucnym (2)
| Pewne | Możliwe |
Aminoreks
Fenfluramina
Deksfenfluramina
Toksyczny olej rzepakowy
Benfluoreks
Inhibitory zwrotnego wychwytu serotoniny | Kokaina
Fenylopropanolamina
Ziele dziurawca
Chemioterapeutyki
Interferon a i b
Leki amfetaminopodobne |
| Prawdopodobne | Mało prawdopodobne |
Amfetaminy
L-tryptofan
Metamfetaminy
Dasatynib | Doustne leki antykoncepcyjne
Estrogeny
Dym papierosowy |
Z grupy 1 wydzielono dwie patologie o nieco odmiennym od PAH charakterze zmian patomorfologicznych w krążeniu płucnym: zarostową chorobę żył płucnych (ang. pulmonary veno-occlusive disease – PVOD) i/lub naczyniakowatość płucną kapilarną (ang. pulmonary capillary haemangiomatosis – PCH) oraz przetrwałe nadciśnienie płucne noworodków.
Grupa 2 dotyczy przypadków nadciśnienia płucnego w wyniku niewydolności skurczowej lub rozkurczowej lewej komory oraz wad zastawkowych serca. Zgodnie z ostatnimi zaleceniami, grupa ta została poszerzona o PH w przebiegu wrodzonych i nabytych kardiomiopatii oraz wrodzonych i nabytych zaburzeń napływu i odpływu z lewej komory serca (2). Jest to tak zwane pozawłośniczkowe (postkapilarne, żylne) nadciśnienie płucne, które przebiega z podwyższonym ciśnieniem zaklinowania w tętnicy płucnej (> 15 mmHg). W praktyce klinicznej ten typ PH rozpoznawany jest często na podstawie współistnienia echokardiograficznych cech nadciśnienia płucnego i obniżonej frakcji wyrzutowej lewej komory serca.
Grupa 3 dotyczy przypadków PH w przebiegu chorób płuc i/lub hipoksji, przebiegających z upośledzeniem rezerw wentylacyjnych typu obturacyjnego albo restrykcyjnego, a ponadto: zaburzeń oddychania w czasie snu, zaburzeń wentylacji pęcherzykowej, długotrwałego przebywania na dużej wysokości oraz wad wrodzonych układu oddechowego (1).
Grupa 4 to przypadki zakrzepowo-zatorowego nadciśnienia płucnego (ang. chronic thromboembolic pulmonary hypertension – CTEPH), które rozwija się u 0,1-9,1% chorych po ostrym epizodzie zatoru tętnicy płucnej (5). Autorzy zajmujący się epidemiologią CTEPH zwracają uwagę na fakt, że 40-60% jego przypadków nie jest poprzedzone ostrym epizodem zatorowości płucnej i mogą one sprawić szczególną trudność diagnostyczną (5). Czynniki ryzyka rozwoju CTEPH to między innymi: duża objętość skrzepliny, nieadekwatna antykoagulacja, przewlekły stan zapalny, choroba nowotworowa, choroby mieloproliferacyjne, stan po splenektomii, zakażone cewniki dożylne/elektrody stymulatora, leczenie substytucyjne z powodu niedoczynności tarczycy, krążące przeciwciała antykardiolipinowe, podwyższone stężenie czynnika VIII w osoczu (6).
Grupa 5 gromadzi przypadki nadciśnienia płucnego o mnogich lub niejasnych przyczynach. Należy tu między innymi PH w przebiegu: sarkoidozy, histiocytozy płucnej z komórek Langerhansa, limfangioleiomiomatozy, zaburzeń metabolicznych, chorób układu krwiotwórczego, po splenektomii, w przebiegu włókniejącego zapalenia śródpiersia (1). Zgodnie z zaleceniami Kongresu w Nicei, do grupy tej przeniesiono (z grupy 1) nadciśnienie płucne w przebiegu przewlekłej niedokrwistości hemolitycznej, z uwagi na jego złożoną patogenezę (2).
Diagnostyka nadciśnienia płucnego
Wstępne podejrzenie nadciśnienia płucnego ustalane jest na podstawie: wywiadu, objawów klinicznych, badania radiologicznego klatki piersiowej i badania elektrokardiograficznego. Objawami przedmiotowymi PH są: upośledzenie tolerancji wysiłku, duszność wysiłkowa i omdlenia wysiłkowe, niekiedy z towarzyszącym bólem zamostkowym (7). W badaniu przedmiotowym stwierdzane są: wzmożenie drugiego tonu serca nad tętnicą płucną i szmer skurczowy niedomykalności zastawki trójdzielnej. Objawy niewydolności prawej komory: wypełnione żyły szyjne, obrzęki kończyn dolnych, powiększenie wątroby, wodobrzusze, przemawiają za obecnością zaawansowanego nadciśnienia płucnego (7).
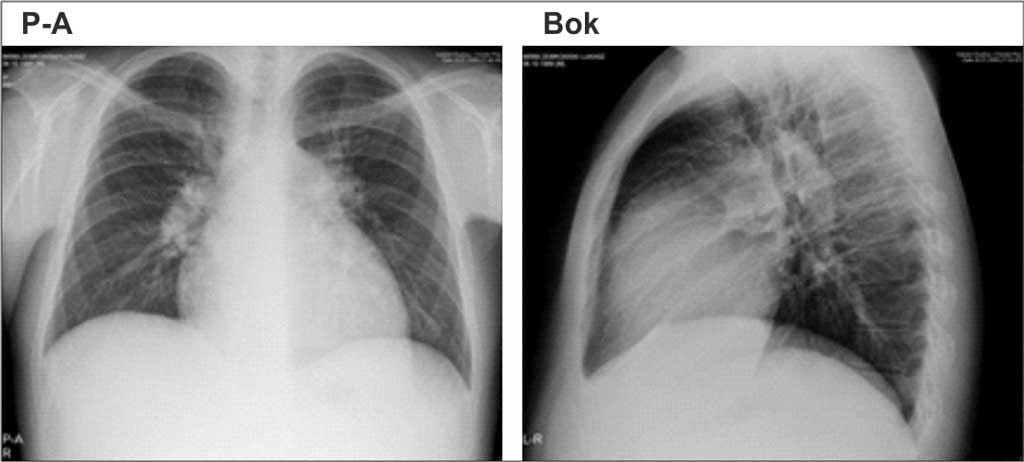
W radiogramie klatki piersiowej za rozpoznaniem PH przemawiają: poszerzenie proksymalnych części tętnic płucnych z amputacją ich odcinków obwodowych, poszerzony pień tętnicy płucnej, powiększona sylwetka serca w projekcji tylno-przedniej ze zwiększonym polem przylegania prawej komory do mostka w projekcji bocznej lewej (2). Objawy PH w badaniu radiologicznym klatki piersiowej przedstawiono na rycinie 2. Objawy te mogą być trudne do oceny u chorych ze zmianami miąższowymi w płucach, szczególnie gdy towarzyszy im powiększenie węzłów chłonnych śródpiersia.
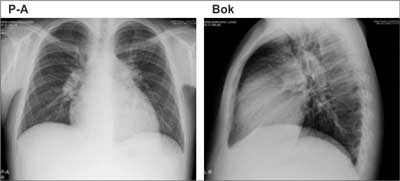
Ryc. 2. Badanie radiologiczne klatki piersiowej u chorego na idiopatyczne tętnicze nadciśnienie płucne – powiększona sylwetka serca, poszerzony pień tętnicy płucnej oraz naczynia płucne we wnękach
W badaniu elektrokardiograficznym, u chorych na PH stwierdza się prawogram, blok prawej odnogi pęczka Hisa, P-pulmonale, cechy przerostu i/lub przeciążenia prawej komory, jednak są to objawy świadczące o zaawansowanym okresie choroby.
Podstawowym badaniem służącym do weryfikacji wstępnego podejrzenia PH jest echokardiografia przezklatkowa (1). Podstawowym parametrem mierzonym podczas badania echokardiograficznego jest maksymalna prędkość fali zwrotnej przez zastawkę trójdzielną. Dodatkowo oceniane są trzy kategorie objawów: A – związanych z prawą komorą, B – związanych z tętnicą płucną, C – związanych z żyłą główną dolną i prawym przedsionkiem. Istotne jest, aby występowały objawy z co najmniej dwóch różnych kategorii. Algorytm oceny prawdopodobieństwa PH w oparciu o wynik badania echokardiograficznego, według Europejskiego Towarzystwa Kardiologicznego, przedstawiono w tabeli 2. W niektórych sytuacjach klinicznych echokardiografia przezklatkowa ma ograniczoną wartość w rozpoznawaniu nadciśnienia płucnego. Ma to miejsce między innymi w przypadku trudności w uwidocznieniu fali zwrotnej przez zastawkę trójdzielną (problem ten dotyczy 1/3 chorych z patologią płucną) oraz u chorych z krążeniem hiperkinetycznym, kiedy to prędkość fali zwrotnej przez zastawkę trójdzielną nie zawsze koreluje z wysokością nadciśnienia płucnego (7).
Tabela 2. Prawdopodobieństwo nadciśnienia płucnego według badania echokardiograficznego
| Maks. prędkość fali zwrotnej przez zastawkę trójdzielną (m/s) | Obecność innych echokardiograficznych objawów PH | Prawdopodobieństwo PH według badania echokardiograficznego |
| ≤ 2,8 | Nie | Niskie |
| ≤ 2,8 | Tak | Pośrednie |
| 2,9-3,4 | Nie | Pośrednie |
| 2,9-3,4 | Tak | Wysokie |
| > 3,4 | Niewymagane | Wysokie |
Tomografia komputerowa (ang. computed tomography – CT) w opcji naczyniowej (angio-CT) może mieć pomocnicze znaczenie w ocenie obecności nadciśnienia płucnego. Cechy nadciśnienia płucnego w badaniu angio-CT to: powiększenie prawej komory serca, poszerzenie (> 29 mm) pnia tętnicy płucnej, zwiększenie współczynnika szerokości tętnicy płucnej do aorty powyżej 1. Devaraj i wsp. wykazali, że łączna ocena badania echokardiograficznego i angio-CT pozwala na uzyskanie lepszej korelacji wyników z badaniem hemodynamicznym (8).
W ostatnich latach pojawiło się wiele doniesień dotyczących roli rezonansu magnetycznego serca (ang. cardiac magnetic resonance imaging – CMRI) w diagnostyce nadciśnienia płucnego. Badanie to, z uwagi na koszt i ograniczoną dostępność, nie jest wykorzystywane rutynowo do rozpoznawania PH. Jednak podkreślana jest jego przydatność do oceny morfologii i funkcji prawej komory serca u chorych na PAH, a także do monitorowania czynności prawej komory u chorych na PAH poddanych leczeniu celowanemu (9, 10).
Parametrem o znaczeniu pomocniczym w diagnostyce PH jest pomiar stężenia mózgowego peptydu lub propeptydu natriuretycznego (ang. brain natriuretic peptide – BNP/ang. N-terminal pro-brain natriuretic peptide – NT-proBNP) (11, 12). Bodźcem do jego syntezy jest mechaniczne rozciąganie jam serca (11, 12). Podwyższone stężenie NT-proBNP lub BNP wskazuje na kardiogenną przyczynę duszności u chorych podejrzanych o PH, jednak nie różnicuje pomiędzy prawo- a lewokomorową niewydolnością serca (13). Stężenie peptydów natriuretycznych wzrasta z wiekiem, dlatego też ustalenie granicy wartości prawidłowej jest trudne. Według Williamsa i wsp. stężenie NT-proBNP > 395 pg/ml pozwalało na prognozowanie PH u chorych na twardzinę układową z 56% czułością i 95% swoistością (14).
Podejrzenie nadciśnienia płucnego ustalone na podstawie badania echokardiograficznego wymaga rozważenia wskazań do cewnikowania prawego serca. Mając na uwadze, że cewnikowanie serca jest badaniem inwazyjnym, wskazania do tego badania powinny być ograniczone do sytuacji, w których wynik cewnikowania pozwoli na wdrożenie odpowiedniego leczenia lub ma istotne znaczenie rokownicze. Zgodnie z obecnym stanem wiedzy, cewnikowanie prawego serca jest zalecane w następujących sytuacjach klinicznych:
– podejrzenie PAH, ocena efektu leków działających na naczynia płucne stosowanych w PAH,
– podejrzenie wrodzonych wad przeciekowych serca,
– u chorych z PH w przebiegu schyłkowej choroby płuc lub serca, kwalifikowanych do przeszczepienia,
– przy podejrzeniu PH u chorego z patologią płucną lub chorobą lewego serca, kiedy jest to pomocne w diagnostyce różnicowej.
Diagnostyka różnicowa nadciśnienia płucnego
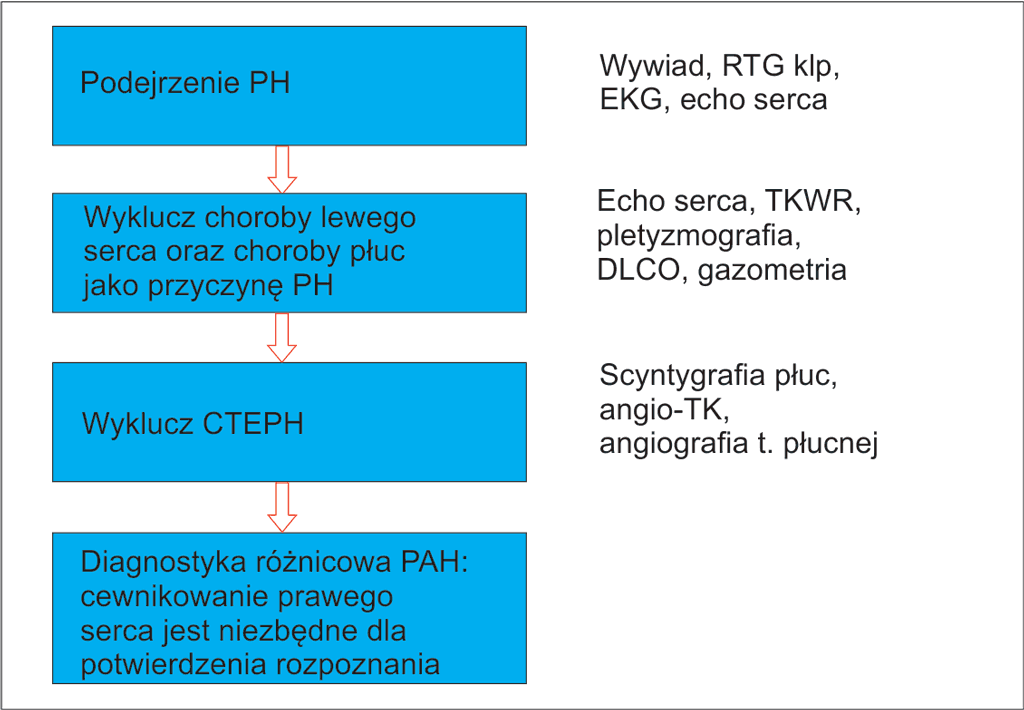
W algorytmie diagnostycznym PH (ryc. 3) Europejskie Towarzystwo Kardiologiczne zaleca rozważenie w pierwszej kolejności najczęstszych populacyjnych przyczyn PH, a więc niewydolności lewej komory (grupa 2) i choroby płuc i/lub hipoksji (grupa 3) (3).
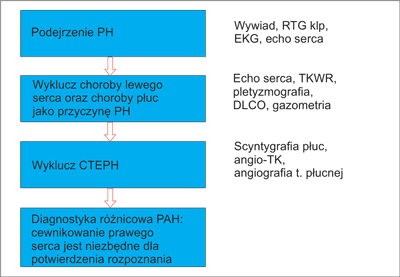
Ryc. 3. Diagnostyka różnicowa nadciśnienia płucnego
Żylne nadciśnienie płucne
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Galie N, Humbert M, Vachiery J-L et al.: 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J 2015; 46: 879-882.
2. Simmoneau G, Gatzoulis MA, Adatia I et al.: Updated clinical classification of pulmonary hypertension. JACC 2013; 62(25), suppl. D: 34-41.
3. Hoeper MM, Bogaard HJ, Condliffe R et al.: Definitions and diagnosis of pulmonary hypertension. JACC 2013; 62(25), suppl. D: 42-50.
4. McGoon MD, Benza RL, Escribano-Subias P et al.: Pulmonary arterial hypertension. Epidemiology and registries. JACC 2013; 62(25), suppl. D: 51-59.
5. Lang IM, Pesavento R, Bonderman D et al.: Risk factors and basic mechanisms of chronic thromboembolic pulmonary hypertension: a current understanding. Eur Respir J 2013; 41: 462-468.
6. Moraca RJ, Kanwar M: Chronic thromboembolic pulmonary hypertension. Heart Fail Clin 2012; 8: 475-483.
7. Noordegraaf AV, Bogaard HJ, Groeneveldt JA et al.: Pulmonary hypertension: diagnosis, differential diagnosis and pitfalls. Eur Respir Monograph 2012; 57: 17-25.
8. Devaraj A, Wells AU, Meister MG et al.: Detection of pulmonary hypertension with multidetector CT and echocardiography alone and in combination. Radiology 2010; 254: 609-616.
9. Benza RL, Biederman R, Murali S, Gupta H: Role of cardiac magnetic resonance imaging in the management of patients with pulmonary arterial hypertension. JACC 2008; 52: 1683-1692.
10. Mc Lure LER, Peacock AJ: Cardiac magnetic resonance imaging for the assessment of the heart and pulmonary circulation in pulmonary hypertension. Eur Respir J 2009; 33: 1454-1466.
11. Thakkar V, Stevens W, Prior D et al.: The inclusion of N-terminal pro-brain natriuretic peptide in a sensitive screening strategy for sclerosis-related pulmonary arterial hypertension: a cohort study. Arthrit Res Ther 2013; 15: R193.
12. Fijałkowska A, Kurzyna M, Torbicki A et al.: Serum N-terminal brain natriuretic peptide as a prognostic parameter in patients with pulmonary hypertension. Chest 2006; 129: 1313-1321.
13. Mahadavan G, Nguyen TH, Horowitz D: Brain natriuretic peptide: a biomarker for all cardiac disease? Curr Opin Cardiol 2014; 29: 160-166.
14. Williams MH, Handler CE, Akram R et al.: Role of N-terminal brain natriuretic peptide (NT-proBNP) in scleroderma-associated pulmonary arterial hypertension. Eur Heart J 2006; 27: 1485-1494.
15. Thenappan T, Shah SJ, Gomberg-Maitland M et al.: Clinical characteristics of pulmonary hypertension in patients with heart failure and preserved ejection fraction. Circ Heart Fail 2011; 4: 257-265.
16. Vachiery J-L, Adir J, Barbera JA et al.: Pulmonary hypertension due to left heart diseases. JACC 2013; 62(25), suppl. D: 100-108.
17. Seeger W: Pulmonary hypertension in chronic lung diseases JACC 2013; 62(25), suppl. D: 109-116.
18. Behr J, Ryu JH: Pulmonary hypertension in interstitial lung disease. Eur Respir J 2008; 31: 1357-1367.
19. Nathan SD, Shlobin OA, Ahmad S et al.: Pulmonary hypertension and pulmonary function testing in idiopathic pulmonary fibrosis. Chest 2007; 131: 657-663.
20. Modrykamien AM, Gudavalli R, McCarthy K, Parambil J: Echocardiography, 6-minute walk distance and distance-saturation product as predictors of pulmonary arterial hypertension in idiopathic pulmonary fibrosis. Respir Care 2010; 55: 584-588.
21. Zisman DA, Karlamangla AS, Kawut SM et al.: Validation of a method to screen for pulmonary hypertension in advanced idiopathic pulmonary fibrosis. Chest 2008; 133: 640-645.
22. Cottin V, Le Pavec J, Prevot G et al.: Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur Respir J 2010; 35: 105-111.
23. Mejia M, Carillo G, Rojas-Serrano J et al.: Idiopathic pulmonary fibrosis and emphysema. Decreased survival associated with severe pulmonary arterial hypertension. Chest 2009; 136: 10-15.
24. Chaouat A, Bugnet AS, Kadaoui N et al.: Severe pulmonary hypertension and chronic obstructive lung disease. Am J Respir Crit Care Med 2005; 172: 189-194.
25. Thabutt G, Dauriat G, Stern JB et al.: Pulmonary hemodynamics in advanced COPD candidates for lung volume reduction surgery or lung transplantation. Chest 2005; 127: 1531-1536.
26. Weitzenblum E, Chaouat A, Kessler R: Pulmonary hypertension in chronic obstructive pulmonary disease. Pneumonol Alergol Pol 2013; 81: 390-398.
27. Lettieri CJ, Nathan SD, Barnett SD et al.: Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest 2006; 129: 746-752.
28. Shorr AF, Wainright JL, Cors CS et al.: Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J 2007; 30: 715-721.
29. Baughman RP, Engel PJ, Taylor L et al.: Survival in sarcoidosis associated pulmonary hypertension: the importance of hemodynamic evaluation. Chest 2010; 138: 1078-1085.
30. Hurdman J, Condliffe R, Elliot CA et al.: Pulmonary hypertension in COPD: results from the ASPIRE registry. Eur Respir J 2013; 41: 1292-1301.
31. Załęska M, Błasińska-Przerwa K, Oniszh K et al.: Włóknienie śródpiersia z nadciśnieniem płucnym jako rzadkie powikłanie sarkoidozy. Pneumonol Alergol Pol 2013; 81: 273-280.
32. Fartoukh M, Humbert M, Capron F et al.: Severe pulmonary hypertension in histiocytosis X. Am J Respir Crit Care Med 2000; 161: 216-223.
33. Le Pavec J, Lorillon G, Jais X et al.: Pulmonary Langerhans cell histiocytosis-associated pulmonary hypertension. Clinical characteristics and impact of pulmonary arterial hypertension therapies. Chest 2012; 142: 1150-1157.
34. Cottin V, Harari S, Humbert M et al.: Pulmonary hypertension in lymphangioleiomyomatosis: characteristics in 20 patients. Eur Respir J 2012; 40: 630-640.
35. Kim NH, Delcroix M, Jenkins DP et al.: Chronic thromboembolic pulmonary hypertension. JACC 2013; 62(25), suppl. D: 92-99.
36. Bonderman D, Lang I: Chronic thromboembolic pulmonary hypertension. Eur Respir Monogr 2012; 57: 108-118.
37. Avouac J, Airo P, Meune C et al.: Prevalence of pulmonary hypertension in systemic sclerosis in European Caucasians and metaanalysis of 5 studies. J Rheumatol 2010; 37: 2290-2298.
38. Coghlan JG, Denton CP, Grunig E et al.: Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: the DETECT study. Ann Rheum Dis 2014; 73: 1340-1349.
39. Le Pavec J, Girgis RE, Lechtzin N et al.: Systemic sclerosis – related pulmonary hypertension associated with interstitial lung disease. Arthrit Rheum 2011; 63: 2456-2464.
40. Launay D, Sitbon O, Hachulla E et al.: Survival in systemic sclerosis-associated pulmonary hypertension in the modern management era. Ann Rheum Dis 2013; 72: 1940-1946.
41. Krowka MJ, Rodriguez-Roisin R: Portopulmonary hypertension: a consequence of portal hypertension. Eur Respir Monogr 2012; 97: 58-70.
42. Montani D, Price LC, Dorfmuller P et al.: Pulmonary venoocclusive disease. Eur Respir J 2009; 33: 189-200.
43. Nunes H, Humbert M, Capron F et al.: Pulmonary hypertension associated with sarcoidosis: mechanisms, hemodynamics and prognosis. Thorax 2006; 61: 68-74.
44. Polomis D, Runo JR, Meyer KC: Pulmonary hypertension in interstitial lung disease. Curr Opin Pulm Med 2008; 14: 462-469.
45. Heresi GA, Dweik RA: Sarcoidosis-associated pulmonary hypertension: one size does not fit all. Chest 2009; 135: 1410-1412.
46. Colombat M, Mal H, Groussard O et al.: Pulmonary vascular lesions in end-stage idiopathic pulmonary fibrosis: histopathologic study on lung explant specimens and correlations with pulmonary hemodynamics. Hum Pathol 2007; 38: 60-65.
47. Szturmowicz M, Kacprzak A, Burakowska B et al.: In search of markers of treatment failure and poor prognosis in IPAH – the value of mosaic lung attenuation pattern on thin section CT scans. Mulidiscip Respir Med 2010; 5: 409-416.