© Borgis - Postępy Nauk Medycznych 4/2016, s. 246-252
*Aleksandra Marach, Jarosław Kierkuś, Józef Ryżko, Grzegorz Oracz
Familial adenomatous polyposis in children
Rodzinna polipowatość gruczolakowata u dzieci
Department of Gastroenterology, Hepatology, Feeding Disorders and Pediatrics, The Children’s Memorial Health Institute, Warsaw
Head of Department: prof. Józef Ryżko, MD, PhD
Streszczenie
Rodzinna polipowatość gruczolakowata (FAP) to najczęstszy dziedziczony zespół polipowatości powodowany przez mutacje antyonkogenu APC (ang. adenomatous poliposis coli gene). Mutacje te prowadzą do powstawania nieprawidłowych białek odpowiedzialnych za tworzenie setek polipów gruczalakowatych umiejscowionych w przewodzie pokarmowym, głównie w jelicie grubym, z których nieuchronnie rozwinie się rak jelita grubego. Istnieje kilka wariantów FAP, między innymi poronna postać FAP (AFAP), także powodowana przez mutację w genie APC, która charakteryzuje się mniejszą ilością polipów oraz późniejszym ich rozwojem w porównaniu z klasycznym FAP. Występuje również rodzinna polipowatość gruczolakowata powodowana przez mutację autosomalnie recesywną występującą w genie MUTYH zwana MAP (MUTYH-associated polyposis), której przebieg jest podobny do AFAP. Ryzyko rozwoju złośliwego raka jelita grubego na podłożu gruczolaków jelita grubego sięga niemal 100%, dlatego tak istotny jest uważny i ujednolicony sposób opieki nad pacjentami z FAP. Ze względu na brak polskich wytycznych dotyczących postępowania w przypadku rodzinnej polipowatości gruczolakowatej polscy klinicyści powinni rozważyć wykorzystanie wytycznych opublikowanych przez European Society for Medical Oncology w 2013 roku.
Summary
Familial adenomatous polyposis (FAP) is the most common inherited polyposis syndrome caused by inactivating mutation in the tumor suppressor gene called adenomatous polyposis coli (APC) gene. FAP is characterized by the presence of hundreds of colorectal adenomatous polyps that inevitably lead to colorectal cancer. There are multiple related conditions caused by mutation in APC gene such as attenuated FAP (AFAP) with fewer number of polyps and delayed onset of colon cancer compared to classic FAP and MUTYH-associated polyposis (MAP) which has similar clinical manifestation to AFAP. Risk of malignancy in patients with FAP reaches 100%, that is why careful and standardized clinical management of FAP is required in order to prevent patients from developing malignancies. In the absence of Polish guidelines for the management of familial adenomatous polyposis Polish doctors should consider using Clinical Practice Guidelines on Familial risk-colorectal cancer published in 2013 by European Society for Medical Oncology.

Introduction
Familial adenomatous polyposis (FAP) is a rare autosomal dominant syndrome characterized by the presence of hundreds of gastrointestinal, adenomatous polyps which can develop to colon cancer at early age. FAP is caused by an inactivating mutation in the tumor suppressor gene called adenomatous polyposis coli (APC) gene. At early age symptoms may be absent, but with the development of polyps symptoms may occur. Characteristic for FAP is a large number of polyps (over 100), which usually appear in the second decade of life (1). There are multiple related conditions caused by mutation in APC gene and MUTYH-associated polyposis (MAP) which has similar clinical manifestation. Risk of malignancy in patients with FAP reaches 100%, that is why careful and standardized surveillance is required. Screening investigations include such procedures as genetic testing and colonoscopy. Once the disease is identified, surgery is the best treatment for reduction of colon cancer risk while the role of chemoprevention is limited.
Epidemiology
FAP is the most common inherited polyposis syndrome and occurs in approximately 1:5000 to 1:17,000 (2). The disease occurs de novo with the frequency of somewhere between 1 in 8000 to 1 in 10,000 (3). Among all colorectal cancer FAP make up less than 1% (4).
Diagnosis
To diagnose FAP more than 100 adenomas must be identified in colorectum. Typical for attenuated FAP (AFAP), the milder form of FAP, is the lower number of adenomas and later onset of the disease. Clinical manifestation of MUTYH-associated polyposis (MAP) is very similar to APC-associated AFAP.
Genetics
FAP is caused by mutation in the tumor suppressor gene called adenomatous polyposis coli (APC) gene. APC is located on chromosome 5 in the q21 region and contains 21 exons (5). Mutations in the APC gene are present in 80-90% of patients with FAP (6). In the remaining patients, FAP results from the occurrence of a de novo mutation in the APC gene. The APC gene is responsible for controling β-catenin. When APC gene loss it’s function, β-catenin is active and causes cellular proliferation (7). It leads to increased probability of mutations and accumulation of changes in genetic material. If this occurs, abnormal cells may accumulate and develop into polyps and colon cancer. In case of the recessive form of adenomatous polyposis, known as MYH adenomatous polyposis (MAP) there is mutation in MUTYH gene (fig. 1) (8).
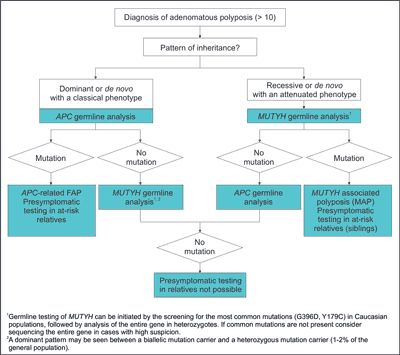
Fig. 1. Algorithm for genetic diagnosis in polyposis syndromes (9)
Signs and symptoms
The majority of pediatric patients with FAP are asymptomatic and undergo evaluation based on their family history (10). At average age of 15 years of age polyps start to develop (11), but at this time symptoms are rare. First signs of FAP occur as polyps start to grow and multiply (tab. 1). It is not safe to wait for symptoms, because it is usually connected with malignant condition. However there have been reported patients who were 5 years old when polyps were diagnosed (12).
Tab. 1. Symptoms of FAP
| Typical | When the disease is advanced |
| Blood in the stool | Continued weight loss |
| Thin stools | Continued lack of energy |
| Diarrhea and/or constipation | |
| Abdominal pain, cramping, or bloating | |
| Anemia | |
There are also such extra-colonic features of FAP present as (tab. 2):
– congenital hypertrophy of the retinal pigment epithelium – CHRPE. CHRPE has been reported to occur in over 50% of the carriers of the mutation in the APC gene (13). It is one of the commonest and earliest extra intestinal manifestations, it lends itself as a screening tool for family members of FAP patients. However the development of molecular biology techniques as well as incomplete penetrance has reduced its significance (14),
– polyps in upper segments of the gastrointestinal tract particularly in stomach and duodenum. Polyps can occur in form of fundus gland polyps or adenomas (15). Stomach polyps occur in approximately 50% of FAP patients and do not have a high potential to form neoplasms. 33% even to 80% of patient with FAP are diagnosed with duodenal polyps (16-18). Polyps located in the ampulla of Vater and the periampullary area can develop into malignant lesion in 4 to 12% of FAP cases (19). The risk of carcinoma is greater in ampullary compared with nonampullary adenomas and increases with the size of adenoma (20),
– dental changes.
Tab. 2. Extra-intestinal features in familial adenomatous polyposis
| Benign lesions | Malignant lesions |
| Congenital hypertrophy of the retinal pigmented epithelium | Duodenal and periampullary adenocarcinoma |
| Epidermoid cysts | Thyroid cancer |
| Osteoma | Brain tumour |
| Desmoid tumour | Hepatoblastoma |
| Supernumerary teeth | |
| Adrenal gland adenomas | |
Supernumerary teeth as well as osteomas and changes in tooth structure:
– desmoid tumours. In about 10% FAP patients non-cancerous tumors most often found in the abdomen are observed (21). It is also proven that desmoid tumours occur 3 times less frequently in men with FAP than in women (22),
– adrenal masses, a benign tumors in adrenal glands,
– osteomas on the skull and jaw and epidermoid cysts – Gardner syndrome.
Patients with FAP have slightly increased risk of malignancies as compared to individuals without FAP:
– duodenal and periampullary adenocarcinomas,
– thyroid cancers are observed in 1% of patients. More than 94% of cases is diagnosed in women (23, 24),
– central nervous system neoplasms (medulloblastoma) occur with low frequency of about 1% and they are usually gliomas. The occurrence of these tumors together with FAP symptoms in the intestine is described as Turcot’s syndrome (25-27),
– hepatoblastomas are rare neoplasms occurring in children. Increased risk of occurrence of these cancers in APC mutation carriers were observed but the incidence did not exceed 1% (28, 29). However it is recommended to check α-fetoprotein levels and hepatic ultrasounds from birth to 5 years of age.
Cancer risks
Patient with FAP who are not diagnosed and treated will develop colon cancer. The average age of malignant transformation diagnosis in untreated patients is 39 years (age 34 to 43 years). Colon cancer occurs by age 20 years in 7%, and by age 25 years in 15% of patients. Extra intestinal malignancies are listed above.
Related conditions
Mutations in the APC gene can cause three related conditions. These conditions are now considered to be variants of familial adenomatous polyposis. These include:
– Gardner syndrome – characterized by the presence of multiple gastrointestinal polyps, osteomas and soft tissue growths (including epidermal cysts, fibromas and desmoid tumors) (30),
– Turcot syndrome – associated with the presence of gastrointestinal polyps and a central nervous system tumor (medulloblastoma) (31),
– attenuated FAP (AFAP) – characterized by fewer polyps (average of about 30 polyps) and delayed onset of colon cancer compared to classis FAP (32).
Researchers recently identified another hereditary form of adenomatous polyposis, MYH-associated polyposis (MAP). Map is caused by a mutation in the MUTYH (also called MYH) gene and is inherited in an autosomal recessive manner. Patients with this disease have fewer polyps (~30) in their colon or rectum than patients with FAP.
Characteristic for MAP is the later age of onset of adenomas and colorectal cancer. Duodenal polyposis seems to be quite frequent and sometimes severe, extraintestinal manifestations such as CHRPE, osteomas and dental anomalies are rarely observed (33).
If any APC pathogenic variant is not identified in patient with colonic polyposis, molecular genetic testing of MUTYH should be considered (34).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Cruz-Correa M, Giardiello FM: Familial adenomatous polyposis. Gastrointest Endosc 2003; 58(6): 885-894.
2. Corredor J, Wambach J, Barnard J: Gastrointestinal polyps in children: advances in molecular genetics, diagnosis, and management. J Pediatr 2001; 138: 621-628.
3. Bisgaard ML, Fenger K, Bülow S et al.: Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat 1994; 3(2): 121-125.
4. Groden J, Thliveris A, Samowitz W et al.: Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991; 66(3): 589-600.
5. Santoro IM, Groden J: Alternative splicing of the APC gene and its association with terminal differentiation. Cancer Res 1997; 57(3): 488-494.
6. Ballantyne GH: Theories of carcinogenesis and their impact on surgical treatment of colorectal cancer. A historical review. Dis Colon Rectum 1988 Jul; 31(7): 513-517.
7. Vogelstein B, Fearon ER, Hamilton SR et al.: Genetic alterations during colorectal-tumor development. N Engl J Med 1988; 319(9): 525-532.
8. Al-Tassan N, Chmiel NH, Maynard J et al.: Inherited variants of MYH associated with somatic G:C T:A mutations in colorectal tumors. Nat Genet 2002; 30(2): 227-232.
9. Balmaña J, Balaguer F, Cervantes A et al.: Familial Risk-Colorectal Cancer: ESMO Clinical Practice Guidelines. Ann Oncol 2013; 24 (suppl. 6): vi73-vi80.
10. Durno CA: Colonic polyps in children and adolescents. Can J Gastroenterol 2007; 21: 233-239.
11. Fearon ER, Vogelstein B: A genetic model for colorectal tumorigenesis. Cell 1990; 61(5): 759-767.
12. Distante S, Nasioulas S, Somers GR et al.: Familial adenomatous polyposis in a 5-year-old child: a clinical, pathological, and molecular genetic study. J Med Genet 1996; 33(2): 157-160.
13. Caspari R, Friedl W, Boker T: Predictive diagnosis in familial adenomatous polyposis: evaluation of molecular genetic and ophthalmologic methods. Z Gastroenterol 1993; 31(11): 646-652.
14. Nusliha A, Dalpatadu U, Amarasinghe B et al.: Congenital hypertrophy of retinal pigment epithelium (CHRPE) in patients with familial adenomatous polyposis (FAP); a polyposis registry experience. BMC Res Notes 2014; 7: 734. doi: 10.1186/1756-0500-7-734.
15. Jagelman DG, DeCosse JJ, Bussey HJ: Upper gastrointestinal cancer in familial adenomatous polyposis. Lancet 1988; 1(8595): 1149-1151.
16. Domizio P, Talbot IC, Spigelman AD et al.: Upper gastrointestinal pathology in familial adenomatous polyposis: results from a prospective study of 102 patients. J Clin Pathol 1990; 43(9): 738-743.
17. Sarre RG, Frost AG, Jagelman DG et al.: Gastric and duodenal polyps in familial adenomatous polyposis: a prospective study of the nature and prevalence of upper gastrointestinal polyps. Gut 1987; 28(3): 306-314.
18. Heiskanen I, Kellokumpu I, Jarvinen H: Management of duodenal adenomas in 98 patients with familial adenomatous polyposis. Endoscopy 1999; 31(6): 412-416.
19. Cordero-Fernández C, Garzón-Benavides M, Pizarro-Moreno A et al.: Gastroduodenal involvement in patients with familial adenomatous polyposis. Prospective study of the nature and evolution of polyps: evaluation of the treatment and surveillance methods applied. European Journal of Gastroenterology and Hepatology 2009; 21: 1161-1167.
20. Basford PJ, Bhandari P: Endoscopic management of nonampullary duodenal polyps. Therapeutic Advances in Gastroenterology 2012; 5: 127-138.
21. Jones IT, Jagelman DG, Fazio VW et al.: Desmoid tumors in familial polyposis coli. Ann Surg 1986; 204(1): 94-97.
22. Klemmer S, Pascoe L, DeCosse J: Occurrence of desmoids in patients with familial adenomatous polyposis of the colon. Am J Med Genet 1987; 28(2): 385-392.
23. Camiel MR, Mulè JE, Alexander LL et al.: Thyroid carcinoma with Gardner’s syndrome. N Engl J Med 1968; 279(6): 326.
24. Bell B, Mazzaferri EL: Familial adenomatous polyposis (Gardner’s syndrome) and thyroid carcinoma. A case report and review of the literature. Dig Dis Sci 1993; 38(1): 185-190.
25. Itoh H, Ohsato K: Turcot syndrome and its characteristic colonic manifestations. Dis Colon Rectum 1985; 28(6): 399-402.
26. Itoh H, Ohsato K, Yao T et al.: Turcot’s syndrome and its mode of inheritance. Gut 1979; 20(5): 414-419.
27. Paraf F, Jothy S, Van Meir EG: Brain tumor-polyposis syndrome: two genetic diseases? J Clin Oncol 1997; 15(7): 2744-2758.
28. Giardiello FM, Petersen GM, Brensinger JD et al.: Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut 1996; 39(96): 867-869.
29. Gruner BA, DeNapoli TS, Andrews W et al.: Hepatocellular carcinoma in children associated with Gardner syndrome or familial adenomatous polyposis. J Pediatr Hematol Oncol 1998; 20(3): 274-278.
30. Gomez Garcia EB, Knoers NV: Gardner’s syndrome (familial adenomatous polyposis): a cilia-related disorder. Lancet Oncol 2009 Jul; 10(7): 727-735. doi: 10.1016/S1470-2045(09)70167-6.
31. Itoh H, Hirata K, Ohsato K: Turcot’s syndrome and familial adenomatous polyposis associated with brain tumor: review of related literature. Int J Colorectal Dis 1993 Jul; 8(2): 87-94.
32. Kory JW, Randall BW: APC-Associated Polyposis Conditions. GeneReviews 1998.
33. Aretz S, Uhlhaas S, Goergens H: MUTYH-associated polyposis: 70 of 71 patients with biallelic mutations present with an attenuated or atypical phenotype. Int J Cancer 2006; 119: 807-814.
34. Sieber OM, Lipton L, Crabtree M et al.: Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med 2003; 348: 791-799.
35. Vogt S, Jones N, Christian D et al.: Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 2009; 137: 1976-1985. e10.
36. Win AK, Cleary SP, Dowty JG et al.: Cancer risks for monoallelic MUTYH mutation carriers with a family history of colorectal cancer. Int J Cancer 2011 Nov 1; 129(9): 2256-2262. doi: 10.1002/ijc.25870.
37. Balmaña J, Balaguer F, Cervantes A, Arnold D: Familial Risk-Colorectal Cancer: ESMO Clinical Practice Guidelines. Ann Oncol 2013; 24 (suppl. 6): vi73-vi80.
38. Bulow S, Bjork J, Christensen IJ et al.: Duodenal adenomatosis in familial adenomatous polyposis. Gut 2004; 53: 381-386.
39. Dekker E, Boparai KS, Poley JW et al.: High resolution endoscopy and the additional value of chromoendoscopy in the evaluation of duodenal adenomatosis in patients with familial adenomatous polyposis. Endoscopy 2009; 41: 666-669.
40. Brosens LA, Keller JJ, Offerhaus GJ et al.: Prevention and management of duodenal polyps in familial adenomatous polyposis. Gut 2005; 54: 1034-1043.
