© Borgis - Nowa Pediatria 3/2016, s. 121-127
Katarzyna Piotrowska1, Piotr Przymuszała1, Bartłomiej Mroziński2, *Katarzyna Jończyk-Potoczna1
Przypadki kliniczne: stwardnienie guzowate u noworodków – interdyscyplinarna jednostka chorobowa
Clinical cases: tuberous sclerosis complex in newborns – interdisciplinary disease
1Studenckie Towarzystwo Naukowe, Zakład Radiologii Pediatrycznej, Uniwersytet Medyczny im. Karola Marcinkowskiego w Poznaniu
Kierownik Zakładu: dr n. med. Katarzyna Jończyk-Potoczna
2Klinika Kardiologii i Nefrologii Dziecięcej, Uniwersytet Medyczny im. Karola Marcinkowskiego w Poznaniu
Kierownik Kliniki: prof. dr hab. n. med. Aldona Siwińska
Summary
Tuberous sclerosis is a rare genetic disease caused by inappropriate differentiation and proliferation of the stem cells, which results in the growth of the hamartomatic tumors in various locations in human body. These may include tumors of the brain, kidneys, heart, skin or eyes, but no pathognomonic sign have yet been found and what is more individual patients may present different combinations of the signs mentioned above. Moreover, these signs may develop in patients at any age, yet some of them, like the rhabdomyomas observed in young children, show strong tendencies to develop on certain stages of the patient’s life. Authors present two clinical cases of the newborns diagnosed with the tuberous sclerosis based on the presence of multiple tumors in their hearts in the cardiac echo and multiple subependymal nodules and subcortical tubers in the magnetic resonance imaging of the central nervous system. In the 24 hours of the Holter monitoring both of the presented newborns presented cardiac arrhythmias, which were successfully treated. Based on the cases described in this article, authors pay attention to the diversity of the symptoms, depending on the organ affected and the age of the final diagnosis, and in turn on the interdisciplinary character of the disease.

Wstęp
Stwardnienie guzowate, inaczej choroba Bourneville’a-Pringle’a (ang. tuberous sclerosis complex – TSC), jest to rzadka, wielonarządowa choroba wynikająca z nieprawidłowego różnicowania i proliferacji komórek macierzystych, powiązana z wielonarządowymi guzami nienowotworowymi typu hamartoma, należąca do grupy fakomatoz. Jest uwarunkowana genetycznie, a u jej podłoża leżą mutacje w genach TSC1 i TSC2, powodujące utratę kontroli nad wzrostem i podziałem komórek, co predysponuje do rozwoju guzów. Nie istnieje jeden patognomoniczny objaw stwardnienia guzowatego. Choroba może być rozpoznawana na podstawie charakterystycznych zmian hamartomatycznych w wielu narządach, takich jak: mózg, skóra, płuca, nerki, oczy czy serce. Zmiany pojawiają się na różnych etapach rozwoju osobniczego, ponadto u poszczególnych chorych określone zmiany mogą nie wystąpić wcale. Jedną z najwcześniej pojawiających się możliwych zmian, bo obserwowalną w badaniu USG już w życiu płodowym, są mięśniaki prążkowanokomórkowe serca, mogące powodować niewydolność serca i arytmie u noworodka, a ustępujące zwykle samoistnie do 2. roku życia dziecka (1). Ich częstość u noworodków ze stwardnieniem guzowatym może wynosić nawet do 90% (2). U każdego noworodka prezentującego tego typu zmiany, w uzupełnieniu diagnostyki różnicowej należy przeprowadzić obrazowanie centralnego układu nerwowego celem poszukiwania również wcześnie pojawiających się guzów podkorowych mózgu. Stwardnienie guzowate jest jednostką wymagającą interdyscyplinarnej opieki i kontroli zarówno guzów już istniejących, jak i pojawiających się w miarę rozwoju, przez lekarzy specjalistów wielu dziedzin medycyny.
Opisy przypadków
Przypadek 1
Pierwsze dziecko, noworodek płci żeńskiej, z ciąży pierwszej porodu pierwszego, urodzony w 38. tygodniu ciąży drogą cięcia cesarskiego z powodu podejrzenia wady serca u płodu i zaburzeń rytmu serca. Przebieg ciąży niepowikłany. Po urodzeniu w badaniu ECHO w Klinice Neonatologii wykluczono wrodzoną wadę serca, natomiast stwierdzono liczne guzy w sercu. U dziecka od urodzenia obserwowano zaburzenia rytmu serca pod postacią przedwczesnych pobudzeń nadkomorowych. Przekazane w 3. tygodniu życia z Kliniki Neonatologii do Kliniki Kardiologii Dziecięcej.
Przy przyjęciu noworodek w stanie ogólnym wyrównanym, bez cech jawnej niewydolności serca. Czynność serca niemiarowa 130-150/min, liczne pojedyncze przedwczesne skurcze dodatkowe. Wątroba niepowiększona, obrzęków obwodowych nie stwierdzono. W wykonanym badaniu echokardiograficznym serca przeprowadzonym w Klinice potwierdzono obecność licznych guzów w sercu (wolna ściana RV 2 guzy o wymiarach: 0,38 x 0,31 cm i 0,5 x 0,4 cm, w dolnej ścianie IVS guz o wymiarach: 0,31 x 0,4 cm, w tylnej ścianie LV 3 guzy o wymiarach: 0,3 x 0,37 cm; 0,33 x 0,33 cm; 0,22 x 027 cm, w górnej części IVS guz o wymiarach: 0,2 x 0,27 cm), bez zaburzeń przepływu krwi przez serce, niewymagających leczenia kardiochirurgicznego. W pierwszym 24-godzinnym badaniu EKG metodą Holtera średnia czynność serca wynosiła 150/min. Stwierdzono pojedynczą ekstrasystolię nadkomorową (7%) i 1/24 h parę pobudzeń nadkomorowych o częstości ośrodka 232/min. Do leczenia włączono propranolol. W kontrolnym 24-godzinnym badaniu EKG metodą Holtera wykazano nasilenie arytmii – 23% pojedynczych przedwczesnych pobudzeń nadkomorowych, 5/24 h par oraz jeden epizod 3 kolejnych pobudzeń nadkomorowych z częstością rytmu 196/min.
Wobec powyższego zdecydowano się na zmianę leczenia antyarytmicznego z propranololu na amiodaron, nie obserwując skutków ubocznych leczenia. W kolejnym kontrolnym 24-godzinnym badaniu EKG metodą Holtera stwierdzono zmniejszenie nasilenia arytmii do 13%, przedwczesnych pojedynczych pobudzeń nadkomorowych, bez par i epizodów częstoskurczu.
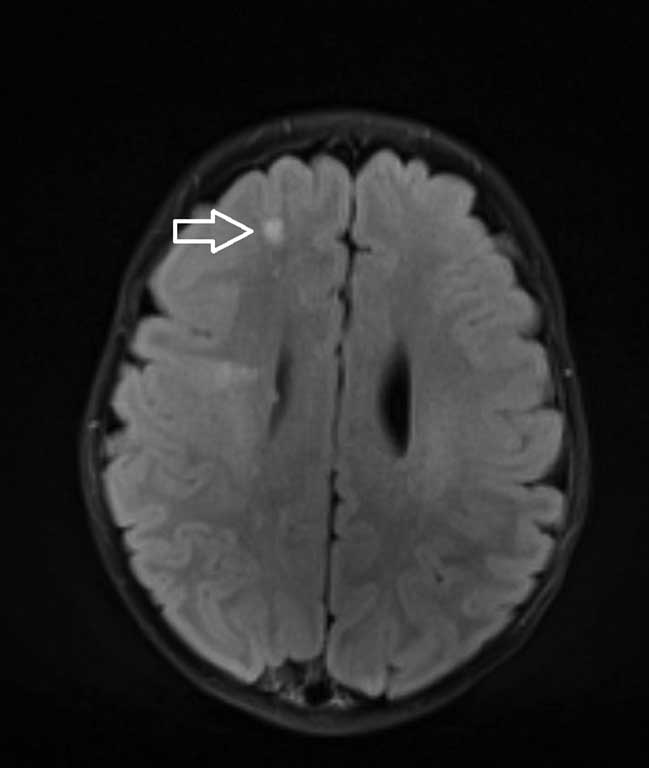
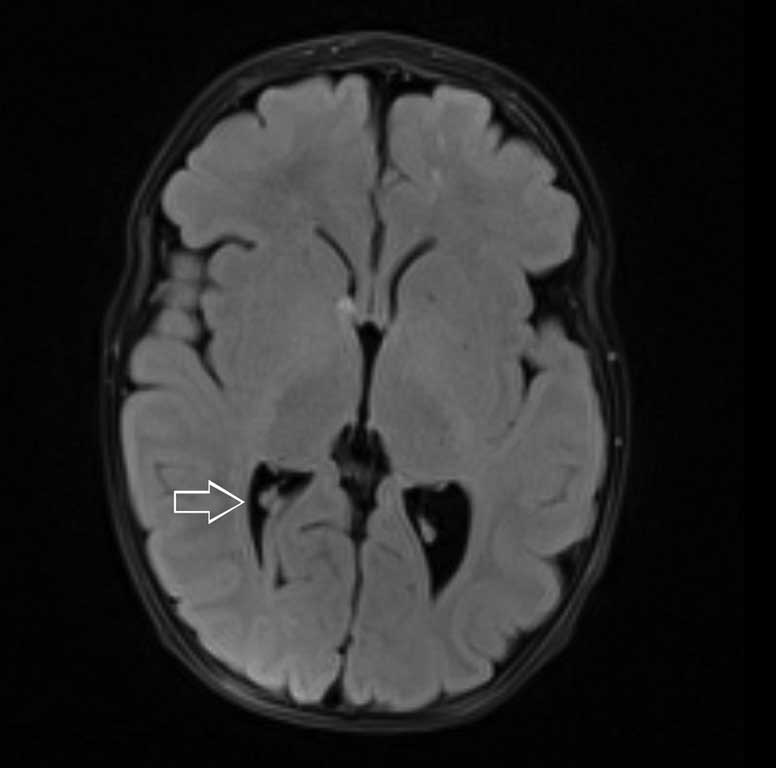
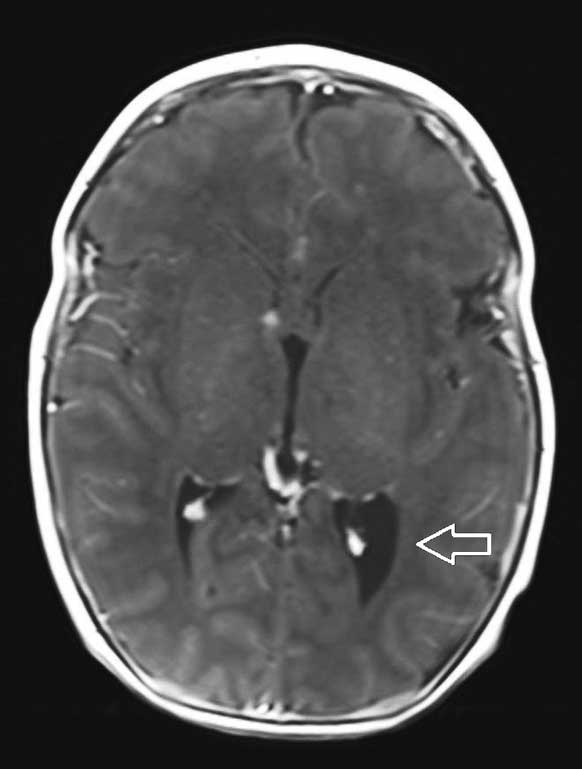
W przeprowadzonym badaniu rezonansu magnetycznego głowy w znieczuleniu ogólnym układ komorowy nieprzemieszczony, poszerzony nieco w zakresie trójkątów komór bocznych. PKB 9 mm, LKB 11 mm, stwierdzono: liczne guzki podwyściółkowe (3-4 mm) w obu półkulach mózgu oraz guzki podkorowe (największe 6 x 5 mm i 5 x 4 mm w prawym płacie czołowym). Po podaniu kontrastu pojedynczy guzek w LKB (poziom trzonu) ulega dyskretnemu wzmocnieniu kontrastowemu. Pozostałe parametry oceniane w badaniu w granicach normy.
Całościowo obraz odpowiadający stwardnieniu guzowatemu (ryc. 1-3).
Ryc. 1. Osiowy obraz MR w sekwencji FLAIR ukazuje guzy podkorowe w postaci hipertensyjnych obszarów zlokalizowanych poniżej zakrętów
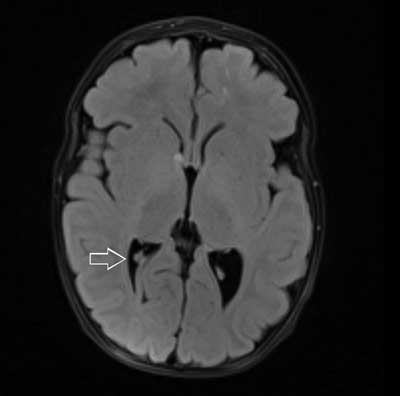
Ryc. 2. Osiowy obraz MR w sekwencji FLAIR ukazuje guzy podwyściółkowe w komorach bocznych mózgu
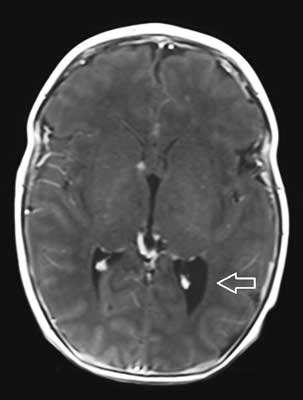
Ryc. 3. Osiowy obraz MR T1-zależny C+ ukazuje guzy podwyściółkowe w komorach bocznych mózgu
Dodatkowo w obrębie ośrodka półowalnego lewego płata czołowego uwidoczniono rozwojową malformację żylną (DVA – z objawem głowy Meduzy poszerzonych żył rdzeniowych, łączących się w żyłę zbiorczą przechodzącą przez korę mózgu) niewymagającą leczenia operacyjnego w ocenie konsultującego neurochirurga.
W wykonanym badaniu EEG zapis czynności bioelektrycznej mózgu w granicach normy wiekowej. W trakcie pobytu u dziecka nie obserwowano zaburzeń neurologicznych. Dziewczynkę zwolniono do domu w stanie ogólnym stabilnym z zaleceniem kontroli w Klinice Neurologii Instytutu Centrum Zdrowia Dziecka w Warszawie.
Przypadek 2
Drugie dziecko, noworodek płci żeńskiej, z ciąży pierwszej porodu pierwszego, urodzony w 39. tygodniu ciąży drogą cięcia cesarskiego z powodu zaburzeń rytmu serca płodu. W prenatalnym badaniu ECHO stwierdzono liczne guzy serca oraz zaburzenia rytmu serca płodu. Po urodzeniu w badaniu ECHO serca w Klinice Neonatologii wykluczono wadę serca oraz potwierdzono obecność licznych guzów w sercu. Od urodzenia obserwowano u noworodka zaburzenia rytmu serca pod postacią licznych pojedynczych przedwczesnych pobudzeń nadkomorowych, okresowo z aberracją przewodzenia śródkomorowego, okresowo układających się w rytm bigeminii i trigeminii nadkomorowej. Przekazany w 3. dobie życia z Kliniki Neonatologii do Kliniki Kardiologii Dziecięcej celem dalszej diagnostyki kardiologicznej i leczenia.
Przy przyjęciu noworodek w stanie ogólnym dobrym, bez cech jawnej niewydolności serca, czynność serca niemiarowa 65-140/min, liczne przedwczesne skurcze dodatkowe okresowo zablokowane. Na skórze widoczne zmiany o charakterze plam szagrynowych. W wykonanym w Klinice badaniu ECHO serca potwierdzono obecność licznych guzów w sercu (w LV przy koniuszku: 0,9 x 0,55 cm, przy IVS 0,35 x 0,3 cm, 3 guzy o wymiarach 0,3 x 0,2 cm, w RV przy koniuszku 3 guzy o wymiarach 0,2 x 0,2 cm, przy IVS 0,2 x 0,2 cm, podnasierdziowo w wolnej ścianie LV: 1,3 x 0,6 cm) bez zaburzeń przepływu krwi w sercu, niewymagających leczenia kardiochirurgicznego.
W pierwszym 24-godzinnym badaniu EKG metodą Holtera stwierdzono średnią czynność serca 88/min oraz bardzo liczne pojedyncze przedwczesne pobudzenia nadkomorowe okresowo układające się w bigeminię zablokowaną, która dominuje w zapisie, okresowo przewiedzione z abberacją przewodzenia śródkomorowego. Dodatkowo zarejestrowano również pary pobudzeń i jeden epizod trzech kolejnych pobudzeń nadkomorowych o częstości ośrodka 232/min. Włączono do leczenia amiodaron, stwierdzając stopniowe zmniejszenie się nasilenia stopnia arytmii.
W kontrolnym 24-godzinnym badaniu EKG metodą Holtera zarejestrowano około 10% pojedynczych przedwczesnych pobudzeń nadkomorowych okresowo zablokowanych, okresowo z abberacją przewodzenia śródkomorowego, oraz kilkanaście par pobudzeń nadkomorowych o częstości ośrodka 197/min. Nie rejestrowano epizodów częstoskurczu, utrzymano leczenie amiodaronem.
W badaniach laboratoryjnych podwyższone stężenie bilirubiny całkowitej oraz nieznaczne podwyższenie wskaźnika BNP. Obserwowano normalizację stężenia BNP.
W przeprowadzonym badaniu rezonansu magnetycznego głowy w znieczuleniu ogólnym układ komorowy nieprzemieszczony, poszerzony nieco w zakresie trójkątów komór bocznych, PKB 9 mm, LKB 7 mm, stwierdzono: liczne guzki podwyściółkowe (3 x 5 mm) w obu komorach bocznych oraz podkorowe, największy w lewej półkuli mózgu (15 x 11 mm i 9 x 9 mm w lewym płacie ciemieniowym) oraz drobne w lewym i prawym płacie czołowym. Po podaniu kontrastu bez cech wzmocnienia.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Dobrzańska A, Ryżko J: Pediatria. Podręcznik do Lekarskiego Egzaminu Końcowego i Państwowego Egzaminu Specjalizacyjnego. [W:] Jóźwiak S, Gołębiewska M (red.): Wybrane zagadnienia neurologii dziecięcej. Wyd. II. Elsevier Urban & Partner, Wrocław 2014: 961-962. 2. Jóźwiak S, Kawalec W, Dłuzewska J: Cardiac tumours in tuberous sclerosis: their incidence and course. Eur J Pediatr 1994; 153(3): 155-157. 3. Kliegman RM, Stanton BF, St. Geme JW et al.: Nelson Textbook of Pediatrics. [In:] Sahin M (ed.): Neurocutaneous syndromes. 19th ed. Elsevier Saunders, Philadelphia 2011: 2049-2050. 4. Barkovich AJ, Koch BL, Moore KR: Diagnostic Imaging: Pediatric Neuroradiology. [In:] Vezina G (ed.): Multiple Regions, Brain. 2nd ed. Elsevier, Philadelphia 2015: 10-13. 5. Park SH, Pepkowitz SH, Kerfoot C et al.: Tuberous sclerosis in a 20-week gestation fetus: immunohistochemical study. Acta Neropathol 1997; 94(2): 180-186. 6. Crino PB, Nathanson KL, Henske EP: The tuberous sclerosis complex. N Engl J Med 2006; 355(13): 1345-1356. 7. Roach ES, Gomez MR, Northrup H: Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 1998; 13(12): 624-628. 8. Hinton R, Parkash A, Romp RL et al.: Cardiovascular Manifestations of Tuberous Sclerosis Complex and Summary of the Revised Diagnostic Criteria and Surveillance and Management Recommendations From the International Tuberous Sclerosis Consensus Group. J Am Heart Assoc 2014; 3(6): e001493. 9. Geva T, Santini F, Pear W et al.: Cardiac rhabdomyoma. Rare cause of fetal death. Chest 1991; 99(1): 139-142. 10. Schwartz AR, Fernandez G, Kotulska K et al.: Tuberous sclerosis complex: advances in diagnosis, genetics and management. J Am Acad Dermatol 2007; 57: 189. 11. Yates JRW: Tuberous sclerosis. Eur J Hum Genet 2006; 14: 1065-1073.