© Borgis - Medycyna Rodzinna 4/1999, s. 22-27
Kamil Hozyasz, Grażyna Rowicka
Cholestazy u dzieci. Patofizjologia, diagnostyka i leczenie
z Kliniki Pediatrii Instytutu Matki i Dziecka
Kierownik Kliniki: prof. dr hab. med. Andrzej Milanowski

Wątroba stanowi 5% masy ciała noworodka i 2% osoby dorosłej (1200-1600 g). Jest największym narządem jamy brzusznej. Podstawową jednostką morfologiczno-czynnościową wątroby jest gronko, tworzone przez komórki skupione wokół końcowych odgałęzień żyły wrotnej i tętnicy wątrobowej (unaczynienie czynnościowe i odżywcze, całkowita ilość krwi przepływającej przez gruczoł wynosi ok. 25% pojemności minutowej serca), kanalików żółciowych, naczyń limfatycznych i nerwów. 60% objętości żółci jest wytwarzane przez hepatocyty i wydzielane do pierwotnych kanalików żółciowych, utworzonych przez wyspecjalizowaną część błony komórkowej biegunów wydzielnicznych sąsiadujących ze sobą komórek. Cholangiocyty – komórki nabłonkowe dróg żółciowych, stanowiące tylko 3-5% populacji komórek wątroby, wydzielają pozostałe 40% objętości żółci – dostarczają do niej wodę i jony. Procesy wydzielnicze, zachodzące w tych niejednorodnych czynnościowo komórkach, znajdują się pod ścisłą kontrolą hormonalną, np. sekrecję żółciowych wodorowęglanów pobudza sekretyna a hamuje somatostatyna. Cholangiocyty są komórkami aktywnymi immunologicznie, często dochodzi do ich uszkodzenia w chorobie przeszczep przeciw gospodarzowi. Kanaliki żółciowe zespalają się ze sobą, tworząc po licznych połączeniach przewód wątrobowy prawy i lewy. Z połączenia w obrębie wnęki wątroby obu przewodów wątrobowych powstaje przewód wątrobowy wspólny, który po krótkim przebiegu poniżej wątroby oddaje wsteczny przewód – przewód pęcherzykowy i dalej biegnie jako przewód żółciowy wspólny do dwunastnicy. Linią podziału dróg żółciowych na wewnątrz- i zewnątrzwątrobowe jest ich instrumentalna dostępność.
Termin cholestaza obejmuje patologiczne procesy spowodowane obniżoną syntezą i wydzielaniem żółci do dwunastnicy, zależne od:
– anatomicznego i/lub czynnościowego utrudnienia odpływu żółci,
– pierwotnego uszkodzenia komórek wątrobowych.
Klasycznie cholestazami zewnątrzwątrobowymi nazywa się cholestazy powodowane pierwotnie przez patologie dróg żółciowych zlokalizowane poniżej miejsca wyjścia prawego i lewego przewodu wątrobowego.
Poznano liczne mechanizmy prowadzące do rozwoju cholestazy wewnątrzwątrobowej:
– zaburzenia dopływu substancji odżywczych do komórek wątrobowych i zmniejszenie aktywności transportu błonowego, np. we wstrząsie, niewydolności krążenia,
– zwiększenie przepuszczalności okołokomórkowej, np. w cholestezie ciężarnych,
– zaburzenia regulacji wydzielania żółci, np. w cholestazie indukowanej limfokinami – aktywacja kinazy białka C (PKC), we wstrząsie – zwiększenie wewnątrzkomórkowego stężenia jonów wapnia,
– zanikanie przewodów żółciowych prowadzące do upośledzenia odpływu żółci, co powoduje: precypitację micelli,
– zmiany polarności komórki wątrobowej i utrata zdolności transportu cząsteczek zjonizowanych, np. w niedokrwieniu, zamknięciu dróg żółciowych,
– uszkodzenia elementów szkieletowych komórek wątrobowych: dysfunkcja mikrokanalików obniżająca sprawność transportu w obrębie hepatocytów (np. w trakcie leczenia chloropromazyną), zmiany struktury mikrowłókienek podporowych powodujące brak kurczliwości bieguna wydzielniczego hepatocytów, np. w niedokrwieniu.
Gromadzące się u pacjentów z cholestazą kwasy żółciowe wywierają bezpośrednie toksyczne działanie na komórki wątrobowe.
Podstawowymi objawami klinicznymi cholestazy są: żółtaczka o różnym nasileniu, świąd skóry, powiększenie wątroby (czasem także i śledziony), odbarwione stolce, ciemniejący mocz, rzadziej żółtaki skórne. Najczęstszymi nieprawidłowościami w zakresie badań laboratoryjnych są: hiperbilirubinemia związana [przy bilirubinie całkowitej powyżej 34 µmol/L = 2 mg/dl – bilirubina związana (bezpośrednia) stanowi powyżej 20%; w surowicy stwierdza się obecność bilirubiny δ, tj. frakcji sprzężonej bilirubiny związanej siłami kowalentnymi z albuminami], wydłużony czas protrombinowy, obecność lipoproteidu X (Lp-x, powstaje u pacjentów z fosfolipidemią), podwyższone stężenia w surowicy: kwasów żółciowych, fosfatazy alkalicznej (izoenzymu wątrobowego), transaminaz, GGTP, cholesterolu, miedzi (ryc. 1).
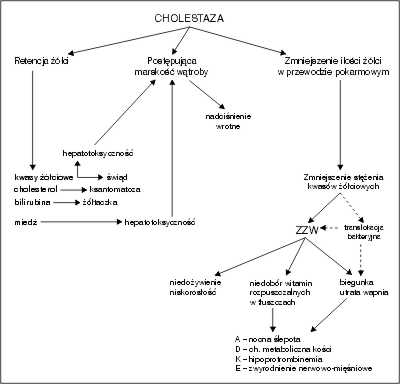
Ryc. 1. Podstawowe skutki cholestazy. Według P.F. Whitington, Pediatr. Clin. North. Am. 1996, 43, 1-26.
Stopień podwyższenia stężenia bilirubiny i kwasów żółciowych, jak i ich wzajemne relacje są bardzo zmienne. Zdarza się, że chorzy zgłaszający się do lekarza z powodu świądu (kumulujące się kwasy żółciowe podrażniają włókna nerwowe przewodzące czucie bólu) bez towarzyszącej żółtaczki/hiperbilirubinemii są długotrwale leczeni objawowo bez przeprowadzenia diagnostyki zmierzającej do wykluczenia chorób wątroby. Stężenia bilirubiny całkowitej w surowicy krwi <= 1 mg% (<= 17,2 µmol/L) oraz bilirubiny związanej <= 0,3 mg% uważa się za prawidłowe. Poza okresem noworodkowym istnieje duża rezerwa wydzielnicza bilirubiny zabezpieczająca przed cholestazą, przy przeciętnej produkcji tego barwnika z rozpadłych erytrocytów wynoszącej ok. 4 mg/kg m.c./dobę – zdolność wydzielania bilirubiny z żółcią sięga 55 mg/kg m.c./dobę.
W pierwszej dobie życia stężenia bilirubiny całkowitej nie powinny przekraczać 5 mg%, a granicą prawidłowego wzrostu stężenia w okresie noworodkowym jest 12,9 mg% dla noworodków donoszonych i 15 mg% dla wcześniaków, przy czym stężenia bilirubiny związanej nie powinny przekraczać 1,5-2 mg%. Przyjmuje się, że kliniczne objawy żółtaczki fizjologicznej mogą trwać nie dłużej niż 7-10 dni u noworodków donoszonych i 14 dni u wcześniaków.
Pierwotne kwasy żółciowe są wytwarzane z cholesterolu w hepatocytach. Wtórne kwasy żółciowe powstają przy udziale flory bakteryjnej z pierwotnych kwasów żółciowych, nie wchłoniętych w jelicie krętym (kwas cholowy -> kwas dezoksycholowy, kwas chenoddezoksycholowy -> kwas litocholowy). Stężenia całkowite kwasów żółciowych w surowicy oznacza się na czczo (< 5 µmol/L) i po posiłku (2-3-krotna zwyżka).
Metabolizm kwasów żółciowych (k.ż.) w okresie noworodkowym charakteryzuje się:
– niewydolnością resorpcji k.ż. w jelicie krętym,
– niskimi wartościami klirensu wątrobowego k.ż. z krążenia wrotnego (zdrowe noworodki mają „podwyższone” stężenia k.ż. w surowicy),
– o połowę mniejszą całkowitą pulą k.ż. w porównaniu z osobami dorosłymi,
– sprzęganiem k.ż. głównie z tauryną.
Oznaczenie stężenia kwasów żółciowych jest czulszym testem w wykrywaniu cholestaz niż bilirubiny. Brak obecności kwasów żółciowych w treści dwunastniczej przemawia za niedrożnością dróg żółciowych. W żółtaczkach czynnościowych, jak choroba Gilberta i Dubin-Johnsona, wykazują wartości prawidłowe.
Część składników stałych żółci (na które przypada 2-3% jej ciężaru) podlega krążeniu wątrobowo-jelitowemu). Należą do nich sole i barwniki żółciowe, cholesterol. U zdrowego człowieka wydalone ze stolcem w ciągu doby kwasy żółciowe stanowią ok. 20% ich całkowitej puli. Urobilinogen, będący produktem bakteryjnej przemiany bilirubiny w jelitach, jest w połowie resorbowany i z krążeniem wrotnym dociera do wątroby, skąd jest ponownie wydzielany z żółcią. Tylko niewielka ilość urobilinogenu dociera do dużego krążenia i jest wydalana przez nerki (u dorosłych ok. 4 mg/dobę).
Nie ma ścisłych granic określających tolerancję komórek wątrobowych na przedłużającą się cholestazę. Uważa się, że szczególnie aktywnej diagnostyki wymaga cholestaza okresu noworodkowego i wczesnoniemowlęcego, z powodu szybko postępujących nieodwracalnych zmian destrukcyjnych w obrębie wątroby i dróg żółciowych. U niemowląt nawet jednomiesięczny zastój żółci może prowadzić do żółciowej marskości wątroby. Niezdiagnozowane choroby metaboliczne, które mogą być przyczyną cholestazy, powodują niedorozwój psychoruchowy dzieci, któremu w wielu przypadkach można zapobiec, stosując np. odpowiednią dietę eliminacyjną. U pacjentów pediatrycznych szczególnie w okresie niemowlęcym, bardzo trudno jest różnicować cholestazę wewnątrz- i zewnątrzwątrobową na podstawie nieprawidłowości w badaniach biochemicznych. Zwykle wysokie (> 600 U/L) stężenia GGTP i fosfatazy alkalicznej stwierdza się w atrezji dróg żółciowych i innych przypadkach niedrożności dróg żółciowych, zespołach skąpości wewnątrzwątrobowych dróg żółciowych, niedoborach α1-antytrypsyny. Wysokie stężenia fosfatazy alkalicznej i niskie GGTP obserwuje się w postępujących rodzinnych cholestazach i wrodzonych zaburzeniach syntezy kwasów żółciowych. Jednocześnie występują zwykle niskie lub średnio podwyższone stężenia powyższych enzymów w chorobach pierwotnie dotyczących hepatocytów, jak np. idiopatyczne noworodkowe zapalenie wątroby. Stężenia kwasów żółciowych i bilirubiny związanej nie są również szczególnie użyteczne w diagnostyce różnicowej cholestaz. W postępujących rodzinnych cholestazach wewnątrzwątrobowych i wrodzonych zespołach skąpości wewnątrzwątrobowych dróg żółciowych stężenia kwasów żółciowych są znacząco podwyższone, natomiast stężenia bilirubiny są podwyższone tylko w niewielkim stopniu. Wysokie stężenia aminotransferaz mogą wskazywać na uszkodzenie hepatocytów. Do postawienia ostatecznego rozpoznania zwykle niezbędne jest wykonanie diagnostyki obrazowej (USG, CT, MR, scyntygrafia wątroby i dróg żółciowych, cholangiografia) oraz badań histopatologicznych.
Diagnostyka różnicowa cholestaz obejmuje bardzo wiele chorób, które ze względu na etiologię można pogrupować następująco: choroby metaboliczne, abberacje chromosomalne (np. zespół Downa), zakażenia, toksemie, cholestazy wewnątrzwątrobowe przetrwałe, cholestezy wewnątrzwątrobowe nawracające, torbielowatości wątroby (ch. Caroliego, torbielowatość mnoga typu dziecięcego), choroby pozawątrobowe, inne przyczyny (wstrząs, hipoperfuzja, choroby rozrostowe, choroby związane z niedrożnością lub zapaleniem jelit, noworodkowy liszaj rumieniowaty).
Choroby metaboliczne
Zaburzenia metabolizmu aminokwasów:
– tyrozynemia – przejściowa tyrozynemia noworodka zwykle ustępuje samoistnie w pierwszym miesiącu życie; tyrozynemia typu I – charakterystyczne są wysokie stężenia α-fetoproteiny przy zasadniczo prawidłowych wynikach testów wątrobowych, podstawą rozpoznania jest obecność w surowicy i moczu bursztynyloacetooctanu i bursztynyloacetonu, w hodowli fibroblastów skóry stwierdza się obniżoną aktywność hydrolazy furnaryloacetooctanu. Poprawę może przynieść zastosowanie NTBC (inhibitor jednego z enzymów szlaku degradacji tyrozyny) i diety z ograniczeniem fenyloalaniny, tyrozyny i metioniny,
– metioinemia.
Zaburzenia metabolizmu węglowodanów:
– galaktozemia – przyczyną może być deficyt jednego z trzech enzymów biorących udział w metabolizmie galaktozy, przy czym najczęściej występuje niedobór urydylilotransferazy galaktozo-1-fosforanu, powodujący najcięższą odmianę choroby tzw. galaktozemię klasyczną. W obrazie klinicznym dominują: objawy uszkodzenia wątroby – hiperbilirubinemia, skaza krwotoczna, uogólnione obrzęki, zaburzenia ze strony oun, zakażenia, zaćma. Diagnostyka opiera się na oznaczaniu aktywności enzymu w krwinkach czerwonych. W leczeniu stosuje się dietę eliminacyjną – ścisłą bezmleczną. U pacjentów z galaktozemią przestrzegających diety stwierdza się wyższe stężenie metabolitów galaktozy w porównaniu z osobnikami zdrowymi, co powodowane może być przez endogenne wytwarzanie toksycznych metabolitów oraz obecność galaktozy w produktach roślinnych. Pomidory, papaja i daktyle zawierają ponad 10 mg tego cukru w 100 g,
– fruktozemia,
– zaburzenia spichrzania glikogenu.
Zaburzenia metabolizmu lipidów:
– choroba Wolmana,
– choroba Gauchera,
– choroba Niemanna-Picka.
Zaburzenia metabolizmu kwasów żółciowych (powodowane niedoborem dehydrogenazy 3β-hydroksysteroidowej, 5β-reduktazy-delta-4,3-oksosteroidowej, 24,25-dihydroksycholanowego enzymu rozszczepiającego. Stanowią przyczynę ok. 3-5% postępujących wewnątrzwątrobowych cholestaz klasyfikowanych jako „idiopatyczne”. Charakteryzują się one prawidłowymi lub niskimi stężeniami k.ż. w surowicy, obecnością nieprawidłowych metabolitów k.ż. w moczu, GGTP w normie, brakiem świądu skóry, zaburzeniami jelitowej absorbcji tłuszczów. Wczesne włączenie leczenia kwasem ursodezoksycholowym (UDCA) może zapobiec marskości wątroby.
Inne zaburzenia metaboliczne:
– mukowiscydoza – obecnie u 5-10% starszych dzieci i młodzieży rozwija się żółciowa marskość wątroby, zagęszczona żółć prowadzi do upośledzenia drożności małych przewodów żółciowych. Leczenie UDCA zmniejsza lepkość żółci, poprawia drenaż żółci, korzystnie działa na homeostazę k.ż. Powyższe leczenie zaleca się u wszystkich pacjentów z dysfunkcją wątroby i dróg żółciowych w przebiegu choroby podstawowej,
– niedobory α1-antytrypsyny – α1-antytrypsyna jest glikoproteiną syntetyzowaną w wątrobie, stanowi ok. 80% α1-globulin surowicy krwi i odpowiada za 80-90% aktywności antyproteazowej osocza. Obraz kliniczny niedoborów jest wynikiem braku równowagi układu proteazy-antyproteazy (rozedma płuc, zapalenie tkanki podskórnej, cholestaza, marskość wątroby). W surowicy stwierdza się obniżone stężenia α1-globulin, w bioptatach wątroby obecne są kuliste wtręty w obrębie cytoplazmy barwiące się PAS-dodatnio. α1-antytrypsyna kodowana jest przez ponad 20 różnych alleli, z których tylko niektóre związane są z tworzeniem nieaktywnych inhibitorów proteaz. Dostępna jest diagnostyka fenotypu i genotypu, przy czym najczęstszym genotypem związanym z chorobą jest homozygotyczny genotyp PiZ. Tylko u niewielu pacjentów rozwija się marskość wątroby, a jedynym skutecznym leczeniem pozostaje przeszczep wątroby,
– choroba Wilsona (dziedziczy się autosomalnie recesywnie z częstością 1/30 000, poznano ponad 40 mutacji lokusu 13q14.3, z których najczęstszą jest WND – występuje w ok. 40% przypadków. Pierwotnym zaburzeniem jest upośledzenie wydzielania miedzi z hepatocytów do kanalików żółciowych. Biochemiczne nieprawidłowości występują od urodzenia, objawy kliniczne zaczynają ujawniać się po skończeniu 5 r.ż. Obowiązują kryteria rozpoznawcze Sternlieba (co najmniej 2 z 4): 1. obecność brązowych pierścieni Kayser-Fleischera w błonie descementa, 2. objawy neurologiczne, 3. obniżone stężenie ceruloplazminy w osoczu, 4. podwyższona zawartość miedzi w wątrobie. W leczeniu stosuje się środki chelatujące, tiomolibdeniany, cynk, przeszczep wątroby. Mniejsze znaczenie ma stosowanie diety z wyłączeniem produktów zasobnych w miedź (wątróbka, czekolada, orzechy, grzyby, soja, skorupiaki),
– inne stany przeciążenia miedzią,
– zespół Zellwegera – zespół mózgowo-wątrobowo-nerkowy, dziedziczy się autosomalnie recesywnie, występuje z częstością 1/100 000. Podłożem choroby są zaburzenia biogenezy peroksysomów. U pacjentów stwierdza się: zniekształcenia głowy – płaska potylica, płytka krawędź oczodołów, płaska nasada nosa, podniebienie gotyckie, ciężką hipotonię, opóźnienie rozwoju psychoruchowego, ziarniste zwapnienia rzepki i krętarza większego, torbiele kory nerek, hepatomegalię – w mikroskopie elektronowym nie udaje się znaleźć peroksysomów, zgon następuje w 6-12 m.ż.,
– niedoczynność przysadki,
– niedoczynność tarczycy,
– choroba spichrzania żelaza okresu noworodkowego,
– protoporfiria erytropoetyczna (dominującym objawem jest światłowrażliwa skóra, manifestacja wątrobowa choroby występuje w 1-5% przypadków),
– wieloraki niedobór dehydrogenazy acylo-CoA.
Objawami klinicznymi mogącymi sugerować chorobę metaboliczną u dziecka z cholestazą są: hipoglikemia, kwasica metaboliczna, hiperamonemia; nawracające wymioty, zaburzenia wzrastania, dysmorfia; opóźnienie rozwoju psychoruchowego, drgawki, krzywica; zaburzenia czynności serca, „dziwne zapachy”, zaćma.
Zakażenia:
– wirusowe zapalenia wątroby – wzw typu A, B, C; opryszczkowe, CMV, różyczkowe, ospowe, Coxsackie, ECHO, reowirusowe (typ 3), parwowirusowe (typ B19),
– bakteryjne (np. kiła, gruźlica, listerioza),
– pierwotniakowe (np. toksoplazmoza).
Infekcyjne zapalenie wątroby może być, obok samoistnego noworodkowego zapalenia wątroby i skąpości wewnątrzwątrobowych dróg żółciowych, przyczyną zespołu noworodkowego zapalenia wątroby (cholestazy wewnątrzwątrobowej noworodków). Wstępna diagnostyka w podejrzeniu cholestazy noworodkowej powinna obejmować: zebranie wywiadu w kierunku występowania cholestaz w rodzinie i koloru stolców dziecka, badanie przedmiotowe ze szczególnym uwzględnieniem wątroby i śledziony (wielkość, konsystencja), badania w kierunku wad wrodzonych (np. anomalii naczyniowych), badania markerów biochemicznych cholestazy, aminogram surowicy i moczu, test metaboliczny moczu, TSH i T4, chlorki w pocie, pełne opracowanie bakteriologiczne pacjenta i badania serologiczne w kierunku zakażenia HBV i TORCH, diagnostykę obrazową wątroby i dróg żółciowych, biopsję wątroby.
Toksemie:
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
