Katedra i Klinika Pediatrii, Hematologii i Onkologii, Warszawski Uniwersytet Medyczny
Kierownik Kliniki: prof. dr hab. n. med. Michał Matysiak
Niedobór XIII czynnika krzepnięcia jest bardzo rzadką osoczową skazą krwotoczną występującą z częstotliwością jeden na kilka milionów osób (1). Wykrycie niedoboru wymaga wykonania specjalistycznych testów, ponieważ standardowo przeprowadzane w diagnostyce zaburzeń krzepnięcia badania nie wykazują odchyleń od normy. Pierwsze wzmianki w piśmiennictwie dotyczące czynnika XIII pochodzą z połowy XX wieku (1). W 1944 roku Robbins opisał związek między konwersją fibrynogenu do fibryny a obecnością nieznanego osoczowego czynnika (1).
W 1948 roku węgierscy biochemicy Laki i Lorand potwierdzili zależność opisaną przez Robbinsa, a nieznane osoczowe białko zyskało nazwę czynnika stabilizującego fibrynę (FSF). Od nazwisk swoich odkrywców białko nazywane jest także czynnikiem Laki-Loranda (2). Pierwszy przypadek chłopca z krwawieniem spowodowanym niedoborem XIII czynnika krzepnięcia został opisany w międzynarodowym piśmiennictwie przez Duckerta i wsp. w 1960 roku (3). W 1963 roku Międzynarodowy Komitet Mianownictwa Czynników Krzepnięcia Krwi nadał mu nazwę XIII czynnika krzepnięcia.
XIII czynnik krzepnięcia występuje w ustroju jako białko osocza oraz jako postać wewnątrzkomórkowa. Osoczowy czynnik XIII jest protransglutaminazą złożoną z dwóch katalitycznych podjednostek A oraz dwóch regulatorowych podjednostek B połączonych przez wiązania niekowalencyjne (4). Gen kodujący podjednostkę A (F13A1) znajduje się na chromosomie 6 (5). Podjednostkę A wytwarzają głównie komórki szpiku kostnego, monocyty, makrofagi, megakariocyty. Jest ona zbudowana z pięciu części: N-końcowego peptydu aktywacyjnego, domeny rdzeniowej, beta-kanapki, beta-beczułki 1 i beta-beczułki 2. Kluczową częścią podjednostki A jest domena rdzeniowa, w której obrębie znajduje się centrum katalityczne złożone z trzech reszt aminokwasowych – cysteiny 314, histydyny 373, asparaginianu 396. Zlokalizowane jest tu również miejsce wiązania jonów wapnia oraz reszta tryptofanowa odpowiedzialna za stabilizowanie produktu pośredniego. Centrum katalityczne chronione jest przed reagentami przez peptyd aktywacyjny i część beta-beczułki 1 (4, 6-13). Podjednostka B kodowana jest przez gen F13B znajdujący się na chromosomie 1 (14). Ta glikoproteina produkowana jest przez hepatocyty i wydzielana jest przez wątrobę do osocza. Składa się ona z 10 tzw. domen sushi. Podjednostka ta odgrywa kluczową rolę w stabilizowaniu struktury i w transporcie podjednostki A, regulacji aktywności czynnika XIII, polimeryzacji fibrynogenu i tworzeniu wiązań krzyżowych (4, 6-13). W trakcie końcowego etapu procesu krzepnięcia krwi czynnik XIII ulega aktywacji (4, 6-13).
Do zapoczątkowania procesu aktywacji konieczna jest obecność trombiny oraz jonów wapnia. Trombina hydrolizuje wiązanie peptydowe, odszczepiając N-końcowy peptyd aktywacyjny, co osłabia wiązanie pomiędzy podjednostkami A i B. W obecności jonów wapnia i fibryny podjednostki B w postaci dimeru B2 oddysocjowują od podjednostek A. Przemieszczeniu ulega domena beta-beczułki 1, odsłonięte zostaje centrum katalityczne dimeru A2. W ten sposób powstaje zaktywowana forma czynnika XIII będąca transglutaminazą (4, 6-13). Enzym ten katalizuje reakcję, w wyniku której dochodzi do utworzenia wiązania izopeptydowego typu g-glutamylo-ε-lizynowego. Powoduje to sieciowanie łańcuchów α i b fibryny, co w znacznym stopniu wzmacnia właściwości mechaniczne skrzepu (10-13).
Wewnątrzkomórkowa postać XIII czynnika krzepnięcia występuje jako dimer złożony z dwóch podjednostek A. W płytkach wiązanymi substratami są m.in.: miosyna, aktyna, winkulina i filamina biorące udział w procesach adhezji i agregacji płytek (15). W monocytach i makrofagach czynnik XIII pośredniczy w fagocytozie poprzez ułatwianie wiązania receptorów Fcg i receptorów dla składowych układu dopełniacza (16). W eksperymentach przeprowadzonych na modelach mysich wykazano udział dimeru A2 obecnego w leukocytach w gojeniu tkanek (17). W licznych badaniach wykazano również ważną rolę wewnątrzkomórkowej postaci czynnika XIII w warstwie labiryntowej łożyska, gdzie współuczestniczy w wytwarzaniu ścisłych połączeń między macicą i łożyskiem (18, 19).
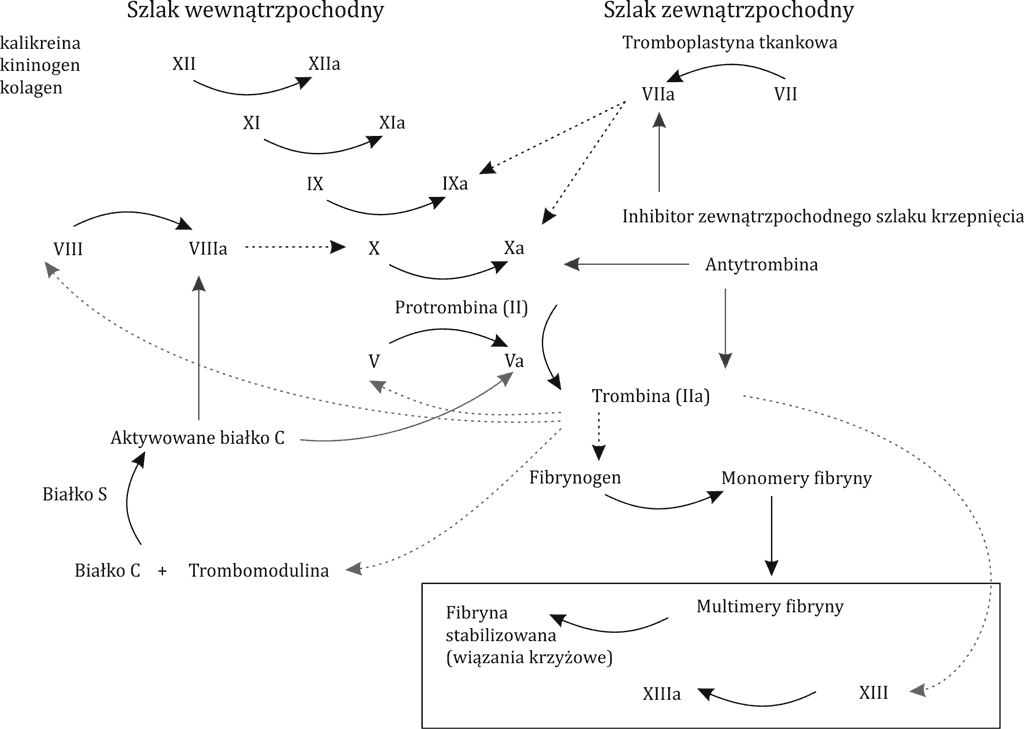
Miejsce działania XIII czynnika w kaskadzie układu krzepnięcia przedstawiono na rycinie 1.
Należy podkreślić, że w przypadku niedoboru czynnika XIII wyniki standardowych testów laboratoryjnych, służących do oceny zaburzeń w układzie krzepnięcia (w tym: INR, PT, APTT, fibrynogen, czas krwawienia), nie odbiegają od normy (21). Pierwszy przypadek tej rzadkiej skazy krwotocznej, opisany przez Duckerta i wsp. w 1960 roku, został wykryty przy pomocy tromboelastografii (3). Tromboelastografia (TEG) pozwalająca na ocenę in vitro dynamiki i jakości procesu tworzenia skrzepu oraz fibrynolizy opracowana została w 1948 roku przez Profesora Helmuta Harterta (22). Wraz z rozwojem techniki powstała jej udoskonalona forma – tromboelastometria (ROTEM) (23). W obu metodach mierzy się zmiany oporów stawianych zanurzonemu we krwi i wykonującemu ruchy rotacyjne trzpieniowi, przez tworzący się lub ulegający lizie skrzep. Wynik przedstawiony jest w formie graficznej tzw. temogramu (23). W przypadku niedoboru czynnika XIII występuje znaczne zmniejszenie maksymalnej amplitudy skrzepu (3).
Historyczną metodą w screeningu niedoboru czynnika XIII jest test zwiększonej rozpuszczalności skrzepu w obecności 5-molowego mocznika lub 2% kwasu chlorooctowego. W warunkach prawidłowych skrzep w roztworze mocznika lub kwasu chlorooctowego powinien pozostać nierozpuszczony przez ponad 24 h, a w przypadku niedoboru czynnika XIII ulega on rozpuszczeniu nawet w ciągu kilku minut (15, 21, 24).
W praktyce w pierwszej kolejności wykorzystuje się dwa testy: test opierający się na pomiarze spektrofotometrycznym ilości uwalnianego amoniaku w czasie aktywacji czynnika XIII lub test inkorporacji znakowanych fluorescencyjnie amin (21, 25, 26).
1. Robbins KC: A study on the conversion of fibrynogen to fibrin. Am J Physiol 1944; 142: 581-588.
2. Laki K, Lorand L: On the solubility of fibrin clots. Science 1948; 108: 280.
3. Duckert F, June E, Schmerling DH: A hitherto undescribed congenital hemorrhagic diathesis probably due to fibrin stabilizing factor deficiency. Thromb Diath Haemorrh 1960; 5: 179-186.
4. McDonagh J: Structure and function of factor XIII. [In:] Colman RW, Hirsh J, Marder VJ, Salzman EW (eds.): Hemostasis and thrombosis: Basic Principles and Clinical Practice. 3rd ed. Lippincott Company, Philadelphia 1994: 301-311.
5. Board PG, Webb GC, McKee J, Ichinose A: Localization of the coagulation factor XIII A subunit gene(F13A) to chromosome bands 6p24-p25. Cytogenet Cell Genet 1988; 48: 25-27.
6. Yee V, Trong I, Bishop P et al.: Structure and function studies of factor XIIIA by x-ray crystallography. Semin Thromb Hemost 1996; 22: 377-384.
7. Pedersen LC, Yee VC, Bishop PD et al.: Transglutaminase factor XIII uses proteinase-like catalytic triad to crosslink macromolecules. Protein Sci 1994; 3: 1131-1135.
8. Ichinose A: The physiology and biochemistry of factor XIII. [In:] Bloom AL, Forbes CD, Thomas DP, Tudenham EGD (eds.): Haemostasis and Thrombosis. Vol. 1. 3rd ed. Livingstone, Edinburgh 1994: 531-546.
9. Bottenus RF, Ichinose A, Davie EW: Nucleotide sequence of the gene tor the B subunit of human factor XIII. Biochemistry 1990; 29: 11195-11209.
10. Siebenlist KR, Meh DA, Mosesson MW: Plasma factor XIII binds specifically to fibrynogen molecules containing gamma chains. Biochemistry 1996; 35: 10448-10453.
11. Greenberg CS, Miraglia CC, Rickles FR et al.: Cleavage of blood coagulation factor XIII and fibrynogen by thrombin during in vitro clotting. J Clin Invest 1998; 75: 1463-1470.
12. Mosesson MW: Fibrinogen gamma chain functions. J Thromb Haemost 2003; 1: 231-238.
13. Moaddel M, Farrell DH, Daugherty MA et al.: Interactions of human fibrinogens with factor XIII: roles of calcium and the gamma peptide. Biochemistry 2000; 39: 6698-6705.
14. Webb GC, Coggan M, Ichinose A, Board PG: Localization of the coagulation factor XIII B subunit gene(F13B) to chromosome bands 1q31-32.1 and restriction fragment length polymorphism at the locus. Hum Genet 1989; 81: 157-160.
15. Hsieh L, Nugent D: Factor XIII deficiency. Haemophilia 2008; 14: 1190-1200.
16. Sarvary A, Szucs S, Balogh I et al.: Possible role of factor XIII subunit A in Fc gamma and complement receptor-mediated phagocytosis. Cell Immunol 2004; 228: 81-90.
17. Matthias N, Sosnovik D, Waterman P et al.: Dual channel optical tomographic imaging of leukocyte recruitment and protease activity in the healing myocardial infract. Circ Res 2007; 100: 1218-1225.
18. Koseki-Kuno S, Yamakawa M, Dickneite G, Ichinose A: Factor XIII A subunit-deficient mice developed severe uterine bleeding events and subsequent spontaneous miscarriages. Blood 2003; 102: 4410-4412.
19. Asahina T, Kobayashi T, Okada Y et al.: Maternal blood coagulation factor XIII is associated with the development of cytotrophoblastic shell. Placenta 2000; 21: 388-393.
20. https://pl.wikipedia.org/wiki/Krzepni%C4%99cie_krwi#/media/File:Coagulation_full.svg.
21. Kohler HPIA, Seitz R, Ariens AS, Muszbek L: On behalf of the factor XIII and fibrynogen SSC Subcommittee of the ISTH diagnosis and classification of factor XIII deficiencies. J Thromb Haemost 2011; 9: 1404-1406.
22. Hartert H: Blutgerninungstudien mit der thromboelastographic, einen neven untersuchingsver fahren. Klin Wochenschr 1948; 16: 257-260.
23. Trzebicki J, Kuźmińska G, Domagała P: Tromboelastometria – nowa metoda wspomagająca decyzje terapeutyczne w zaburzeniach hemostazy, oparta na tromboelastografii Harteta. Pol Mer Lek 2009; 27: 85-91.
24. Jennings I, Kitchen S, Woods TA, Preston FE: Problems relating to the laboratory diagnosis of factor XIII deficiency: a UK NEQUAS study. J Thromb Haemost 2003; 1: 2603-2608.
25. Lewrie AS, Green L, Mackie IJ et al.: Factor XIII – an under diagnosed deficiency – are we using the right assays. J Thromb Haemost 2010; 8: 2478-2482.
26. Ajzner EML: Kinetic spectrophotometric factor XIII activity assays: the subtraction of plasma blank is not omissible. J Thromb Haemost 2004; 2: 2075-2077.
27. Kohler HP, Ichinose A, Seitz R et al.: On behalf of the factor XIII and fibrinogen SCC Subcommitee of the ISTH. J Thromb Haemost 2011; 9: 1404-1406.
28. Karimi M, Bereczky Z, Cohan N, Muszbek L: Factor XIII deficiency. Semin Thromb Hemost 2009; 35: 426-438.
29. De Jager T, Pericleous L, Kokot-Kiereppa M et al.: The burden and management of FXIII deficiency. Haemophilia 2014; 20: 733-740.
30. Anwar R, Miloszewski KJ: Factor XIII deficiency. Br J Haematol 1999; 107: 468-484.
31. Lak M, Peyvandi F, Ali Sharifian A et al.: Pattern of symptoms in 93 Iranian patients with severe factor XIII deficiency. J Thromb Haemost 2003; 1: 1852-1853.
32. Inbal A, Dardik R: Role of coagulation factor XIII in angiogenesis and tissue repair. Pathophysiol Haemost Thromb 2006; 35: 162-165.
33. Ragaz S, Kemp G, Furlan M, Beck EA: Bleeding disorder with abnormal wound healing, acid-soluble clots and normal factor XIII. Thromb Haemost 1976; 36: 537-541.
34. Asahina T, Kobayashii T, Takeuchi K, Kanayama N: Congenital blood coagulation factor XIII deficiency and successful deliveries: a review of the literature. Obstet Gynecol Surv 2007; 62: 255-260.
35. Board PG, Losowsky MS, Miloszewski KJA: Factor XIII: inherited and acquired deficiency. Blood Rev 1993; 7: 229-242.
36. Ichinose A: Pathophysiology and regulation of factor XIII. Thromb Haemost 2011; 86: 57-65.
37. Lorand L, Graham RM: Transglutaminases: crosslinking enzymes with pleotropic functions. Nat Rev Mol Cell Biol 2003; 4: 140-156.
38. Bunn JA: Pathophysiology of Blood Disorders. The McGraw Hill Companies Inc., New York 2016: 2075-2077.
39. Petri M, Ellman L, Carey R: Acquired factor XIII deficiency with chronic myelomonocytic leukemia. Ann Intern Med 1983; 99: 638-639.
40. Luo Y, Zhang G, Zuo W et al.: Acquired factor XIII inhibitor in monocyclonal gammopathy of undeterminated significance: characterization and cross-linked fibrin ultrastructure. Ann Hematol 2010; 89: 833-834.
41. Lorand L, Velasco PT, Hill JM et al.: Intracranial hemorrage in systemic lupus erythematosus associated with an autoantibody against factor XIII. Thromb Haemost 2002; 88: 919-923.
42. Ahmad F, Solymoss S, Poon MC et al.: Characterization of an acquired IgG inhibitor of coagulation factor XIII in a patient with systemic lupus erythematosus. Br J Haematol 1996; 93: 700-703.
43. Henriksson P, Hedner U, Nilsson IM: Factor XIII(fibrin stabilizing factor) in Henoch-Schonlein’s purpura. Acta Pediatr Scand 1977; 66: 273-277.
44. Kamitsuji H, Tani K, Yasui M et al.: Activity of blood coagulation factor XIII as a prognostic indicator in patients with Henoch-Schonlein purpura. Eur J Pediatr 1987; 146: 519-523.
45. Lodemann PKH, Held T, Ivaskevicius V et al.: Acquired deficiency of coagulation factor XIII – possible evidence for a new link between coagulation and infection from a case. Ann Hematol 2013; 92: 427-429.
46. Lopaciuk S, Bykowska K, McDonagh JM et al.: Difference between type 1 autoimmune inhibitors of fibrir stabilization in two patients with severe hemorrhagic disorder. J Clin Invest 1978; 61: 1196-1203.
47. Miesbach WBM, von Auer CH, Scharrer I (eds.): Case report of a FXIII inhibitor in a 77-year-old patient. 34 Hemophilia Symposium. Springer Medizin Verlag, Heidelberg 2004.
48. Otis PT, Feinstein DI, Rapaport SI, Patch MJ: An acquired inhibitor of fibrin stabilization associated with isoniazid therapy: clinical and biochemical observations. Blood 1974; 44: 771-781.
49. Dorgalaleh A, Rashidpanah J: Blood coagulation factor XIII and factor XIII deficiency. Blood Reviews 2016; 30: 461-475.
50. Nugent DJ: Prophylaxis in rare coagulation disorders – factor XIII deficiency. Thromb Res 2006; 118: S23-28.
51. Ichinose A, Asahina T, Kobayshi T: Congenital blood coagulation factor XIII deficiency and perinatal management. Curr Drug Targets 2005; 6: 541-549.
52. Sharief LAT, Kadir RA: Congenital factor XIII deficiency in women: a systematic review of literature. Haemophilia 2013; 19: 349-357.
53. Zawilska K, Chojnowski K, Klukowska A et al.: Polskie zalecenia postępowania w rzadkich niedoborach osoczowych czynników krzepnięcia. Hematologia 2011; 2(4): 303-310.
54. Kerlin BA, Inbal A, Will A et al.: Recombinant factor XIII prophylaxis is safe and effective in young children with congenital factor XIII-A deficiency: international phase 3b trial results. J Thromb Haemost 2017; 15: 1601-1606.
55. Ribizzi G, Farinini D, Gentile R et al.: Factor XIII deficiency and head trauma: management and therapy. Neurol Sci 2015; 36(10): 1933-1934.
56. Ajzner E, Schlammadinger A, Kerenyi A: Severe bleeding complications caused by an autoantibody against the B subunit of plasma factor XIII: a novel form of acquired factor XIII deficiency. Blood 2009; 113: 723-725.
57. Fardella PCG, Gonzalez N, Cuneo M: An acquired inhibitor to factor XIII (case report). Blood 1999; 94: 95b.
58. Krumdieck R, Shaw DR, Huang ST et al.: Hemorrhagic disorder due to an isoniazid-associated acquired factor XIII inhibitor in a patient with Waldenstrom’s macroglobulinemia. Am J Med 1991; 90: 639-645.