*Anna Dusza, Michał Matysiak
Pierwotna małopłytkowość immunologiczna u dzieci
Primary immune thrombocytopenia in children
Katedra i Klinika Pediatrii, Hematologii i Onkologii, Warszawski Uniwersytet Medyczny
Kierownik Kliniki: prof. dr hab. n. med. Michał Matysiak
Summary
In this article we present current investigation on primary immune thrombocytopenia in children. There are described pathomorphology, clinical symptoms, diagnosis and treatment. We also present current data from literature about genetic tests and latest data on treating options in children.
Primary immune thrombocytopenia (ITP) is one of the most frequent hematological disorders in pediatric population. Although the majority of children have a self-limited and short duration of the disease. However, approximately 20-30% of those patients can develop chronic ITP, which can cause significant complications and higher mortality and reduced quality of life. Especially regarding to long-term immunosupression or surgical interventions, such like splenectomy and restrictions on daily activities to avoid trauma. Over the past decades a lot of informations has been reported about pathogenic features of ITP. Nowdays, we know that it is not only caused by increased platet destruction and decreased platet production, but also complex, multifactorial immune dysregulation, like loss of immune tolerance and generation of platelet autoantibodies. In this article we present current investigation on ITP including clinical symptoms, diagnosis, pathomorphology and latests options on treatment in children. We also present current data about genetic biomarker, such as Vanin-1 (VNN-1) which has been suggested as one of predictors of chronic disease and potentially can offer early prognosis estimation.

Definicja
Pierwotna małopłytkowość immunologiczna (ang. primary immune thrombocytopenia – ITP) to choroba, w której dochodzi do izolowanego obniżenia liczby płytek (poniżej 100 000/mm3) we krwi obwodowej, przy braku innych czynników lub zaburzeń mogących powodować trombocytopenię (1).
Jeszcze do niedawna ITP nazywana była „idiopatyczną plamicą małopłytkową”, jednak z uwagi na coraz lepiej poznane immunologiczne tło tej choroby Międzynarodowa Grupa Ekspertów (International Working Group – IWG) w 2009 roku zadecydowała o zmianie nomenklatury odnośnie definicji schorzenia, kryteriów klinicznych oraz odpowiedzi na zastosowane leczenie.
Obecnie głównym kryterium podziału choroby jest czas jej trwania. Na tej podstawie wyróżnia się:
– postać nowo rozpoznaną – do 3 miesięcy trwania choroby (ang. newly diagnosed ITP – ndITP),
– postać przetrwałą – od 3 do 12 miesięcy trwania choroby (ang. persistent ITP – pITP),
– postać przewlekłą – powyżej 12 miesięcy trwania choroby (ang. chronic ITP – cITP).
W większości przypadków choroba przebiega łagodnie i ustępuje samoistnie, najczęściej pomiędzy 2. a 8. tygodniem czasu trwania choroby (2).
Pomimo częstego występowania, choroba wciąż nastręcza problemów terapeutycznych. U około 20-30% pacjentów przechodzi ona w postać przewlekłą (cITP), czyli trwa powyżej roku (3).
Prawdopodobieństwo cITP zwiększa się wraz z wiekiem dziecka w chwili postawienia rozpoznania (4).
U pacjentów powyżej 10. roku życia ryzyko postaci przewlekłej wynosi już 47% (5). Zasadniczą trudnością diagnostyczno-terapeutyczną u pacjentów z ITP jest brak możliwości przewidzenia przebiegu choroby u danego dziecka. To z kolei prowadzi często do obniżenia się jakości życia (ang. quality of life – QoL) u małego pacjenta i jego rodziny, eskalacji strachu i lęku przed nawrotem choroby (6, 7). Niejednokrotnie wiąże się to też z ograniczeniem codziennej aktywności dziecka w celu uniknięcia potencjalnych urazów.
Epidemiologia
Pierwotna małopłytkowość immunologiczna jest najczęstszą skazą płytkową u dzieci. Występuje ona z częstością 1,9-6,4 przypadku na 100 000 dzieci rocznie (8). U dzieci w okresie największej zapadalności na ITP, tj. między 2. a 6. rokiem życia, nie stwierdzono istotnych różnic w częstości występowania choroby pomiędzy płciami (9). Sytuacja zmienia się dopiero w grupie dorastających dziewcząt, gdzie częstość małopłytkowości przechodzącej w postać przewlekłą jest większa.
W pierwotnej małopłytkowości immunologicznej obserwowana jest sezonowość zachorowań. Szczyt przypada na okres jesienno-zimowy, natomiast stosunkowo najmniej nowych przypadków odnotowywanych jest w okresie letnim.
Etiopatogeneza
Dotychczas nie udało się w pełni wyjaśnić wszystkich szlaków doprowadzających do powstania ITP. Najpewniej jej mechanizm jest wielokierunkowy i uzależniony od wielu czynników, w tym predyspozycji genetycznych (10).
Momentem początkowym u dzieci jest zwykle infekcja wirusowa lub szczepienie (zwłaszcza szczepionkami żywymi) (11).
W wyniku pobudzenia układu immunologicznego dochodzi do powstania przeciwciał przeciwpłytkowych, głównie klasy IgG, skierowanych przeciwko glikoproteinom znajdującym się na powierzchni płytek (najczęściej glikoproteinom IIb/IIIa, rzadziej Ib/IX lub innym). Przeciwciała te opłaszczając z jednej strony płytki, łączą się z receptorem Fc gamma na makrofagach, doprowadzając do ich fagocytozy i tym samym do skrócenia czasu przeżycia płytek do 2 dni (12).
Jednocześnie dochodzi do zmniejszenia produkcji płytek, co jest związane z zahamowaniem trombopoezy w szpiku kostnym wskutek uszkodzenia megakariocytów przez przeciwciała przeciwpłytkowe, jak również względny niedobór trombopoetyny (13).
W patogenezie ITP powstaje też szereg zaburzeń dotyczących prawidłowego funkcjonowania układu immunologicznego (14). Dotyczy to m.in. upośledzonej tolerancji limfocytów B, doprowadzając do patologicznego powstania przeciwciał przeciwpłytkowych. Obserwowane są również zaburzenia w komórkach prezentujących antygen (15).
U chorych na ITP zmienia się też profil cytokinowy limfocytów T CD 4+, z przewagą Th0/Th1 przy zmniejszeniu limfocytów Th2 i limfocytów T regulatorowych.
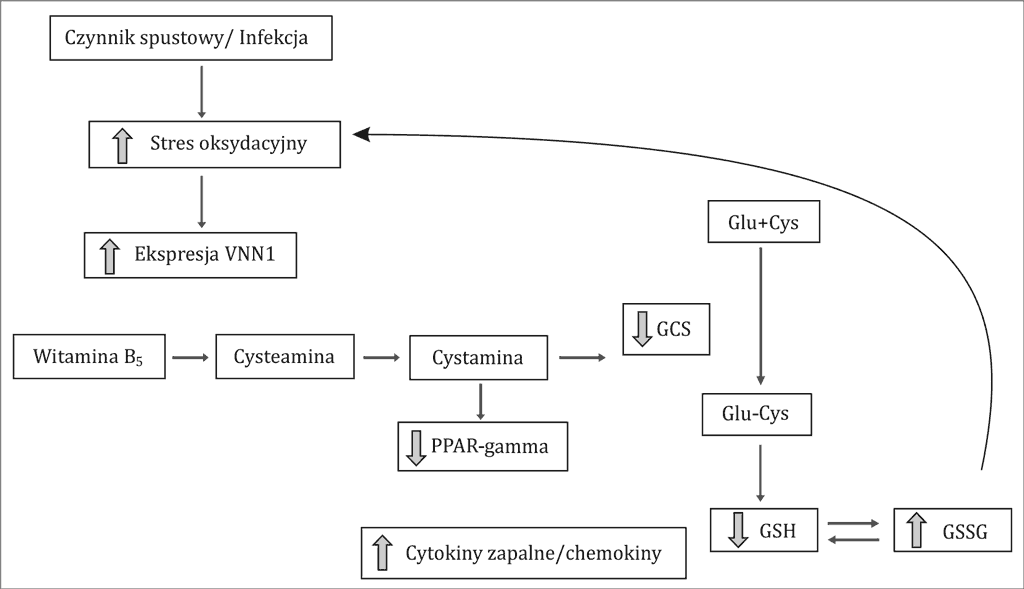
Ostatnio w piśmiennictwie pojawiły się doniesienia przedstawiające rolę nadekspresji genu VNN-1 (Vanin-1) w mechanizmie przewlekłej postaci małopłytkowości (16).
Gen ten opisywany jest jako czujnik procesów oksydoredukcyjnych, a jego nadekspresja wiąże się z niekontrolowanym wzrostem aktywności wolnych rodników. Wzrost stresu tlenowego doprowadza na poziomie molekularnym do destrukcji błon komórkowych i tym samym wiąże się z tworzeniem tzw. neoepitopów, co z kolei stymuluje reakcję immunologiczną skierowaną przeciwko własnym płytkom krwi (ryc. 1).

Ryc. 1. Miejsce VNN1 w szlakach sygnałowych komórek krwi obwodowej w warunkach stresu oksydacyjnego według Berruyer i wsp. (51, 52)
Glu – kwas glutaminowy; Cys – cysteina; GCS – enzym glutamylosyntetaza; GSH – zredukowana forma glutationu; GSSG – utlenowana postać glutationu
W dalszym ciągu trwają jednak badania określające przydatność oznaczenia tego genu u każdego pacjenta z nowo rozpoznaną pierwotną immunologiczną małopłytkowością. W chwili obecnej nie jest to postępowanie wystandaryzowane.
Obraz kliniczny pacjenta z ITP
Najczęstszym objawem pierwotnej małopłytkowości immunologicznej jest skaza krwotoczna, która występuje pod postacią wylewów podskórnych oraz wybroczyn na skórze i śluzówkach całego ciała. Mogą również wystąpić krwawienia z nosa, dziąseł, dróg rodnych i układu moczowego. Ryzyko to wzrasta znacznie przy liczbie płytek < 10 x 109/l (tzw. minimum hemostatyczne), jednak krwawienia w wyniku urazu mogą wystąpić nawet przy większej liczbie płytek (17).
Najbardziej groźne są krwawienia z przewodu pokarmowego i do ośrodkowego układu nerwowego, te ostatnie na szczęście występują stosunkowo rzadko, bo z częstością 0,1-0,5% (18-21).
Nasilenie powikłań krwotocznych może też się zwiększyć z takimi czynnikami ryzyka, jak: wiek chorego, współistniejące zakażenie, niewydolność nerek czy przyjmowane przez pacjenta leki mogące powodować uszkodzenie śluzówki żołądka lub upośledzające hemostazę. W badaniu fizykalnym pacjenci z ITP nie powinni prezentować innych odchyleń od normy poza skazą skórno-śluzówkową. Szczególną czujność diagnostyczną powinny wzbudzić takie niepokojące objawy, jak: uogólniona limfadenopatia, powiększenie wątroby i/lub śledziony, jak również zgłaszane przez pacjenta bóle kostne, poty nocne czy utrata masy ciała.
W takich przypadkach zawsze powinno się wykluczyć choroby limfoproliferacyjne lub też inne schorzenia mogące przebiegać razem z małopłytkowością, np. HIV czy toczeń rumieniowaty układowy (SLE) (22).
Badania diagnostyczne
Rozpoznanie pierwotnej małopłytkowości immunologicznej stawiane jest na zasadzie wykluczenia innych stanów lub chorób (tab. 1).
Tab. 1. Podział małopłytkowości zależny od mechanizmu choroby i jej przyczyny
| Małopłytkowości pierwotne |
| Nabyte | Wrodzone |
Pierwotna małopłytkowość immunologiczna
Małopłytkowość dziecka matki z ITP
Alloimmunologiczna małopłytkowość noworodków | Zespół szarych płytek
Zespół Wiskotta-Aldricha
CAMT |
| Małopłytkowości wtórne |
1. Zaburzenia produkcji:
– zahamowanie czynności szpiku, np. aplazja szpiku, leki, infekcje (PVB19)
– niedobór trombopoetyny
– wyparcie linii trombopoetycznej przez naciek nieprawidłowych komórek nowotworowych, np. białaczka
– nieefektywna trombopoeza, np. niedobór witaminy B12 lub kwasu foliowego |
| 2. Zwiększone niszczenie w mechanizmie immunologicznym, np. SLE, ALPS, choroba Hashimoto, sarkoidoza, miastenia, infekcje |
| 3. Zwiększone zużycie w mechanizmie nieimmunologicznym, np. DIC, sinicze wady serca, naczyniaki, TTP, choroba von Willebranda podtyp 2 B |
| 4. Nieprawidłowa dystrybucja płytek, np. choroba Gauchera, nadciśnienie wrotne |
Kluczowym do postawienia właściwego rozpoznania jest dobrze zebrany wywiad rodzinny i chorobowy.
Obecność małopłytkowości w rodzinie może nasunąć dziedziczne tło choroby i występowanie takich zespołów, jak zespół Wiskotta-Aldricha, choroba von Willebranda typ 2b czy zespół szarych płytek.
W trakcie zbierania wywiadu należy też zwrócić szczególną uwagę na przyjmowane leki, m.in.: niesterydowe leki przeciwzapalne, aspiryna, heparyna, cytostatyki, które mogą również przyczyniać się do powstania małopłytkowości.
Podstawowym badaniem laboratoryjnym wykonywanym u pacjentów z małopłytkowością jest morfologia krwi z oceną liczby retikulocytów oraz rozmazem krwi obwodowej.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Zawilska K: Wstęp do wytycznych diagnostyki i leczenia pierwotnej immunologicznej małopłytkowości. Pol Arch Med Wew 2010; 120: 5-7.
2. Bolton-Maggs PH, Dickerhoff R, Vora AJ: The nontreatment of childhood ITP. Semin Thromb Hemost 2001; 27(3): 269-275.
3. Blanchette V, Bolton-Maggs P: Childhood immune thrombocytopenic purpura: diagnosis and management. Pediatr Clin North Am 2008; 55: 393-420.
4. Donato H, Picon A, Martinez M et al.: Demographic data, natural history, and prognostic factors of idiopathic thrombocytopenic purpura in children: a multicentered study from Argentina. Pediatr Blood Cancer 2009; 52: 491-496.
5. Jackowska T, Wagiel E, Polit A: Pierwotna małopłytkowość immunologiczna w świetle wytycznych American Society of Hematology z 2011 roku. Post Nauk Med 2016; 6: 436-440.
6. Klaassen RJ, Blanchette VS, Barnard D et al.: Validity, reliability, and responsiveness of a new measure of health-related quality of life in children with immune thrombocytopenic purpura: the kids’ ITP Tools. J Pediatr 2007; 150: 510-515.
7. Zilber R, Bortz AP, Yacobovich J et al.: Analysis of health-related quality of life in children with immune thrombocytopenia and their parents using the kids’ ITP tools. J Pediatr Hematol Oncol 2012; 34: 2-5.
8. Terrell DR, Beebe LA, Vesely SK: The incidence of immune thrombocytopenic purpura in children and adults: A critical review of published reports. Am J Hematol 2010; 85(3): 174-180.
9. Neunert C, Lim W, Cohen A et al.: The American Society of Hematology 2011 evidence- based practice guideline for immune thrombocytopenia. Blood 2011; 117: 4190-4207.
10. Zehnder JL, Semple JW, Imbach P et al.: Future research in ITP: an ICIS consensus. Ann Hematol 2010; 89 (suppl. 1): 19-23.
11. Bertuola F, Morando C, Menniti-Ippolito F et al.: Association between drug and vaccine use and acute immune thrombocytopenia in childhood: a case control study in Italy. Drug Saf 2010; 33: 65-72.
12. Podolak-Dawidziak M, Chojnowski K: Małopłytkowość – patofizjologia i klasyfikacja. [W:] Robak T, Warzocha K (red.): Hematologia. Grupa Via Medica, Gdańsk 2016; 400-418.
13. Kaushansky K: Thrombopoetin. N Engl J Med 1998; 339: 746-754.
14. Jernas M, Hou Y, Stomberg Cecling F et al.: Differences in gene expression and cytokine levels between newly diagnosed and chronic pediatric ITP. Blood 2013; 122: 1789-1792.
15. McKenzie CG, Guo L, Freedman J, Semple JW: Cellular immune dysfunction in immune thrombocytopenia (ITP). Br J Haematol 2013; 163(1): 10-23.
16. Zhang B, Lo C, Shen L et al.: The role of vanin-1 and oxidative stress-related pathways in distinguishing acute and chronic pediatric ITP. Blood 2011; 117: 4569-4579.
17. Psaila B, Petrovic A, Page LK et al.: Intracranial hemorrhage (ICH) in children with immune thrombocytopenia (ITP): study of 40 cases. Blood 2009; 114: 4777-4783.
18. Butros LJ, Bussel JB: Intracranial hemorrhage in immune thrombocytopenic purpura: a retrospective analysis. J Pediatr Hematol Oncol 2003; 25(8): 660-664.
19. Zeller B, Rajantie J, Hedlund-Treutiger I et al.: Childhood idiopathic thrombocytopenic purpura in the Nordic Countries: Epidemiology and predictors of chronic disease. Acta Pediatr 2005; 94(2): 178-184.
20. Fogarty P, Segal J: The epidemiology of immune thrombocytopenic purpura. Curr Opin Hematol 2007; 14(5): 515-519.
21. Krivit W, Tate D, White JG et al.: Idiopathic thrombocytopenic purpura and intracranial hemorrhage. Pediatrics 1981; 67(4): 570-571.
22. Neunert C, Lim W, Crowther M et al.: The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011; 117: 4190-4207.
23. Braester A: Pseudothrombocytopenia as pitfall in the treatment of essential thrombocythemia. Eur J Haematol 2003; 70: 251-252.
24. Podolak-Dawidziak M: Diagnostyka pierwotnej małopłytkowości immunologicznej. Pol Arch Med Wew 2010; 120 (suppl.): 8-9.
25. Provan D, Stasi R, Newland AC et al.: International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115: 168-186.
26. Łaguna P, Matysiak M: Diagnostyka i leczenie małopłytkowości u dzieci. Stand Med, Pediat 2015; 12: 992-1002.
27. Veres G, Karoczka I, Bodanszky H et al.: The role of Helicobacter pylori infection in children with chronic immune thrombocythopenic purpura. Orv Hetil 2009; 150: 801-804.
28. Stasi R, Sarpatwrari A, Segal JB et al.: Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood 2009; 113: 1231-1240.
29. Klukowska A: Pierwotna małopłytkowość immunologiczna u dzieci. Pol Arch Med Wew 2010; 120 (suppl.): 25-28.
30. Brighton TA, Evans S, Castaldi PA et al.: Prospective evaluation of the clinical usefulness of an antygen-specific assay (MAIPA) in idiopathic thrombocytopenic purpura ant other immune thrombocytopenias. Blood 1996; 88: 194-201.
31. McMilan R, Wang L, Tani P: Prospective evaluation of immunobead assay for the diagnosis of adult chronic immune thrombocytopenic purpura (ITP). J Thromb Haemost 2003; 1: 485-491.
32. Rodeghiero F, Stasi R, Gernsheimer T et al.: Standarization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113(11): 2386-2393.
33. Rosthoj S, Hedlund-Treutiger I, Rajantie J et al.: Duration and morbidity of newly diagnosed idiopathic thrombocytopenic purpura in children: A prospective Nordic study of an unselected cohort. J Pediatr 2003; 143(3): 302-307.
34. Blanchette V, Carcao M: Approach to the investigation and management of immune thrombocytopeic purpura in children. Semin Hematol 2000; 37: 299-314.
35. Bussel J: Intravenous immune serum globulin in immune thrombocytopenia: clinical results and biochemical evaluation. Vox Sang 1985; 49 (suppl. 1): 44-50.
36. Bussel J, Kimberly RP, Inman RD et al.: Intravenous gammaglobulin treatment of chronic idiopathic thrombocytopenic purpura. Blood 1983; 62: 480-486.
37. Chojnowski K: Leczenie dorosłych chorych na pierwotną małopłytkowość immunologiczną. Pol Arch Med Wew 2010; 120 (suppl.): 10-16.
38. Braendstrup P, Bjerrum OW, Nielsen OJ et al.: Rituximab chimeric anti-CD20 monoclonal antibody treatment for adult refractory idiopathic thrombocytopenic purpura. Am J Hematol 2005; 78(4): 275-280.
39. Bussel JB, Hsieh L, Buchanan R et al.: Long-term use of the thrombopoietin-mimetic romiplostim in children with severe chronic immune thrombocytopenia (ITP). Pediatr Blood Cancer 2015; 62: 208-213.
40. Bussel JB, de Miguel PG, Despotovic JM et al.: Eltrombopag for treatment of children with persistent and chronic immune thrombocytopenia (PETIT) a randomised, multicentre, placebo-controlled study. Lancet Haematol 2015; 2(8): e315-325.
41. Grainger JD, Locatelli F, Chotsampancharoen T et al.: Eltrombopag for children with chronic immune thrombocytopenia (PETIT2): a randomised, multicentre, placebo-controlled trial. Lancet 2015; 386(10004): 1649-1658.
42. Gomez-Almaguer D, Herrera-Rojas MA, Jamie-Perez JC: Eltrombopag and high-dose dexamethasone as frontline treatment of newly diagnosed immune thrombocytopenia in adults. Blood 2014; 123(25): 3906-3908.
43. Neunert CE: Management of newly diagnosed immune thrombocytopenia: can we change outcomes? Blood Adv 2017; 1(24): 2295-2301.
44. Skrobowska-Woźniak A, Rokicka-Milewska R: Wyniki leczenia splenektomią samoistnej skazy płytkowej u dzieci. Ped Pol 1993; 68: 25-30.
45. Aronis S, Platokouki H, Avgeri M et al.: Retrospective evaluation of long-term efficacy and safety of splenectomy in chronic idiopathic thrombocytopenic purpura in children. Acta Pediatr 2004; 93: 638-642.
46. Kuhne T, Blanchette V, Buchnan GR et al.: Splenectomy in children with idiopathic thrombocytopenic purpura. A prospective study of 134 children from the International Childhood ITP Study Group. Pediatr Blood Cancer 2007; 49: 829-834.
47. Togasaki E, Shimizu N, Nagano Y et al.: Long-term efficacy of partial splenic embolization for the treatment of steroid-resistant chronic immune thrombocytopenia. Ann Hematol 2018; 97: 655-662.
48. Kimura F, Itoh H, Ambiru S et al.: Long-term results of initial and repeated partial splenic embolization for the treatment of chronic idiopathic thrombocytopenic purpura. AJR Am J Roentgenol 2002; 179(5): 1323-1326.
49. Davies JM, Barnes R, Milligan D: British Committee for Standards in Hematology, Working Party of the Hematology/Oncology Task Force. Up-date of guidelines for prevention and treatment of infection in patients with an absent or dysfuncional spleen. Ciln Med 2002; 2: 440-443.
50. Provan D, Newland A, Norfolk D et al.: Guidelines for the investigation and management of idiopathic thrombocytopenic purpura in adults, children and in pregnancy. Br J Haematol 2003; 120: 574-596.
51. Sood R, Wong W, Jeng M et al.: Gene expression profile of idiopathic thrombocytopenic purpura. Pediatr Blood Cancer 2006; 47(5 suppl.): 675-677.
52. Sood R, Wong W, Gotlib J et al.: Gene expression and pathway analysis of immune thrombocytopenic purpura. Br J Haematol 2008; 140: 99-103.
53. Smith O: Inherited and congenital thrombocytopenia. Pediatric Hematology 2010; 530: 5-7.
54. Balduini C: Inherited thrombocytopenias: a proposed diagnostic algorithm from the Italian Cruppo di Studio delle Piastrine. Hematologica 2003; 88: 582-592.
55. Lambert MP: What to do when you suspect an inherited platelet disorder. Blood 2011; 1: 377-383.
56. Buchnan GR, Adix L: Grading of hemorrhage in children with idiopathic thrombocytopenic purpura. J Pediatr 2002; 141: 683-688.
57. Bolton-Maggs PH, Moon I: Assessment of UK practice for management of acute childhood idiopathic thrombocytopenic purpura against published guidelines. Lancet 1997; 350: 620-623.