Piotr Kwast1, Olga Olszewska-Sosińska1, Maria Wolniewicz1, Katarzyna Zybert2,3, Dorota Sands2,3, *Lidia Zawadzka-Głos1
Assessment of the severity of chronic sinusitis in children with cystic fibrosis using the Lund-Mackay Score depending on genotype
Ocena ciężkości przewlekłego zapalenia zatok u dzieci z mukowiscydozą w skali Lund-Mackaya w zależności od genotypu
1Department of Paediatric Otolaryngology, Medical University of Warsaw, Poland
Head of Department: Lidia Zawadzka-Głos, MD, PhD
2Cystic Fibrosis Centre, Hospital in Dziekanów Leśny, Poland
Head of Department: Professor Dorota Sands, MD, PhD
3Cystic Fibrosis Department, Institute of Mother and Child, Warsaw, Poland
Head of Department: Professor Dorota Sands, MD, PhD
Streszczenie
Wstęp. Przewlekłe zapalenie zatok dotyczy większości pacjentów z mukowiscydozą (CF). Skala Lund-Mackaya (LM) jest obiektywnym narzędziem pozwalającym w prosty sposób ocenić ciężkość zmian zatokowych w badaniu tomografii komputerowej (TK). Zależności między obrazem klinicznym, wynikami badań obrazowych oraz mutacjami genu CFTR nie są dobrze poznane.
Cel pracy. Określenie korelacji ciężkości zmian w tomografii komputerowej z typem mutacji genu CFTR wywołującej chorobę podstawową u dzieci z mukowiscydozą.
Materiał i metody. Przeanalizowano dane dzieci chorych na mukowiscydozę, u których wykonano badanie TK w latach 2016-2018 w Klinice Otolaryngologii Dziecięcej Warszawskiego Uniwersytetu Medycznego. Wzięto pod uwagę wiek, płeć oraz typ mutacji genu CFTR. Każdą tomografię oceniono w podstawowej i zmodyfikowanej skali Lund-Mackaya (LMS i MLMS). Badanie miało charakter retrospektywny.
Wyniki. Do badania włączono 34 dzieci, 16 dziewczynek i 18 chłopców w wieku od 3 do 17 lat (mediana wieku 10 lat) w momencie wykonania tomografii. Mediana LMS wynosiła 17 (zakres 2-23), a MLMS ? 18 (zakres 2-24), a ich różnica była istotna statystycznie. Szesnaście pacjentów było homozygotami F508del. Ciężką mutację genu CFTR w obu allelach miało 28, a łagodną w co najmniej jednym allelu 6 pacjentów. Mediana LMS w grupie z mutacją ciężką wyniosła 17,5, a w grupie z mutacją łagodną 14,5. Mediana MLMS wyniosła odpowiednio 18 i 14,5. Różnica była istotna statystycznie zarówno dla LMS, jak i MLMS. Nie wykryto istotnej statystycznie korelacji pomiędzy wynikami w skali LMS i MLMS a płcią. Słaba korelacja dodatnia między LMS i MLMS a wiekiem pacjenta nie była istotna statystycznie. U 11 dzieci (32%) stwierdzono hipoplazję lub aplazję jednej lub więcej zatok obocznych nosa.
Wnioski. U dzieci chorych na mukowiscydozę punktacja w zmodyfikowanej skali Lund-Mackaya jest wyższa niż w podstawowej skali LMS. Grupa pacjentów z ciężką mutacją genu CFTR ma wyższe wyniki zarówno LMS, jak i MLMS, niż grupa z mutacją łagodną. Ze względu na częstą u dzieci z mukowiscydozą aplazję zatok, zastosowanie zmodyfikowanej skali Lund-Mackaya pozwala uniknąć błędów podczas porównania wyników TK u różnych pacjentów.
Summary
Introduction. Chronic sinusitis affects most patients with cystic fibrosis (CF). The Lund-Mackay (LM) scale is an objective tool allowing for easy assessment of the severity of lesions in sinuses observed in computed tomography (CT). The link between clinical picture, image results and CFTR gene mutations is not well evaluated.
Aim. To specify the correlation of the severity of lesions found in computed tomography and the CFTR gene mutation type causing an underlying disease in children with cystic fibrosis.
Material and methods. Data of the children with cystic fibrosis who underwent CT between 2016-2018 at the Department of Paediatric Otolaryngology of the Medical University of Warsaw was analysed. The following factors were taken into account: age, sex and CFTR gene mutation type. Each CT was assessed using the basic and modified Lund-Mackay Score (LMS and MLMS). The study was retrospective.
Results. 34 children, 16 girls and 18 boys aged between 3 and 17 years (median age ? 10 years), were enrolled to the study at the time of CT. LMS median was 17 (range 2-23), and MLMS ? 18 (range 2-24), and their difference was statistically significant. 16 patients were F508del homozygous. Severe CFTR mutation in both alleles was observed in 28 patients, and mild mutation was found in at least one allele in 6 patients. LMS median in the group of patients with severe mutation was 17.5, and in the group of patients with mild mutation ? 14.5. MLMS median was 18 and 14.5, respectively. The difference was statistically significant for both LMS and MLMS. No statistically significant correlation was found between the LMS and MLMS results and sex. The weak positive correlation between LMS and MLMS and patient’s age was not statistically significant. Hypoplasia or aplasia of one or more paranasal sinuses was observed in 11 children (32%).
Conclusions. The score on the modified Lund-Mackay scale is higher than the score on the basic LMS scale in children with cystic fibrosis. The group of patients with a severe CFTR gene mutation has higher scores for both LMS and MLMS than the group with a mild mutation. Due to frequent sinus aplasia in children with cystic fibrosis, the use of the modified Lund-Mackay scale allows for error avoidance when comparing CT results of different patients.
Introduction
Cystic fibrosis is a genetic disease affecting approx. 1:4000-5000 persons in Poland (1). It is caused by the mutation of cystic fibrosis transmembrane conductance regulator (CFTR) on chromosome 7 and it is inherited in an autosomal recessive manner. It is estimated that in Poland 1 in 35 people is a carrier of a defective allele. Over 2,000 CFTR gene mutations are currently known, only part of them are responsible for occurrence of CF (1). By means of common screening tests in newborns having been carried out in Poland since 2006, 70-80 new cases of CF are recorded annually (1 per 5750 live births) (1).
Cystic fibrosis is a multi-system disease that mainly involves the respiratory and digestive systems. The increased density and viscosity of mucus in patients contributes to recurrent respiratory infections. Chronic sinusitis is observed in most patients with CF. Some studies report that the disease incidence is determined to be at the level of 100% (2).
The quality of care over the patients with cystic fibrosis in Poland has significantly improved in recent years. However, data regarding median life span in CF patients in Poland and in the West is still different. The average life span is currently over 50 years in Canada (3), while in Poland it equals approx. 35 years (1). Children constitute most patients with CF in Poland.
Complications of chronic bronchopulmonary disease are the most common cause of death in patients with cystic fibrosis (4). Information on the effect of chronic sinusitis on the condition of the lower respiratory tract may be more frequently found in the literature, however this issue needs further studies (5). Early diagnosis and appropriate treatment of chronic inflammation of the upper respiratory tract may be of key importance to keep normal function of lungs.
The clinical course of cystic fibrosis is very variable in the population. Besides the multiplicity of CFTR gene mutations causing the impairment of the chloride channel function in various mechanisms and to a varying degree, the gene expression is also not constant (1). What is more, particular systems and organs are often affected to a varying degree by the disease in individual patients. Due to high variability in the clinical picture of the disease, researchers tend to look for links between the clinical picture, genetic and environmental factors, and treatment results. By means of expansion of the available knowledge on this issue, the life quality and span of patients with cystic fibrosis may be increased. This study focused on the relation between the severity of lesions in the CT of sinuses and the type of mutation causing cystic fibrosis.
Aim
To evaluate the severity of sinus disease using LMS and MLMS in children with CF and find a possible correlation with their genotype.
Material and methods
Data of patients who underwent CT of sinuses between 2016-2018 at the Department of Paediatric Otolaryngology of the Medical University of Warsaw was analysed retrospectively. Information regarding age, sex and genotype of all patients was collected. Aplastic sinuses were recorded in the examined patients. In the event of several tests carried out in one patient, the result of the earliest available test was only considered.
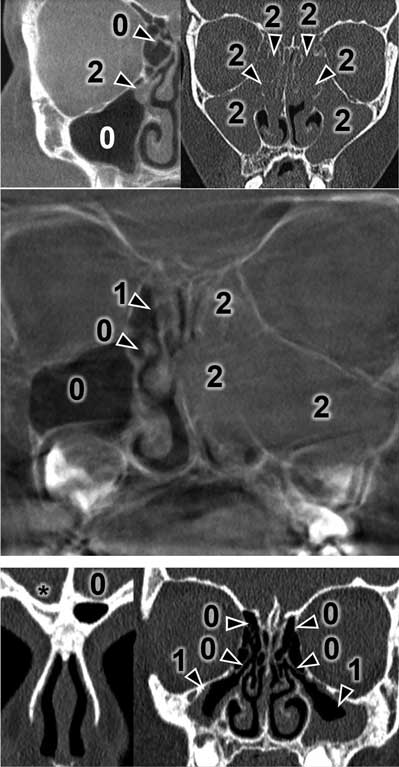
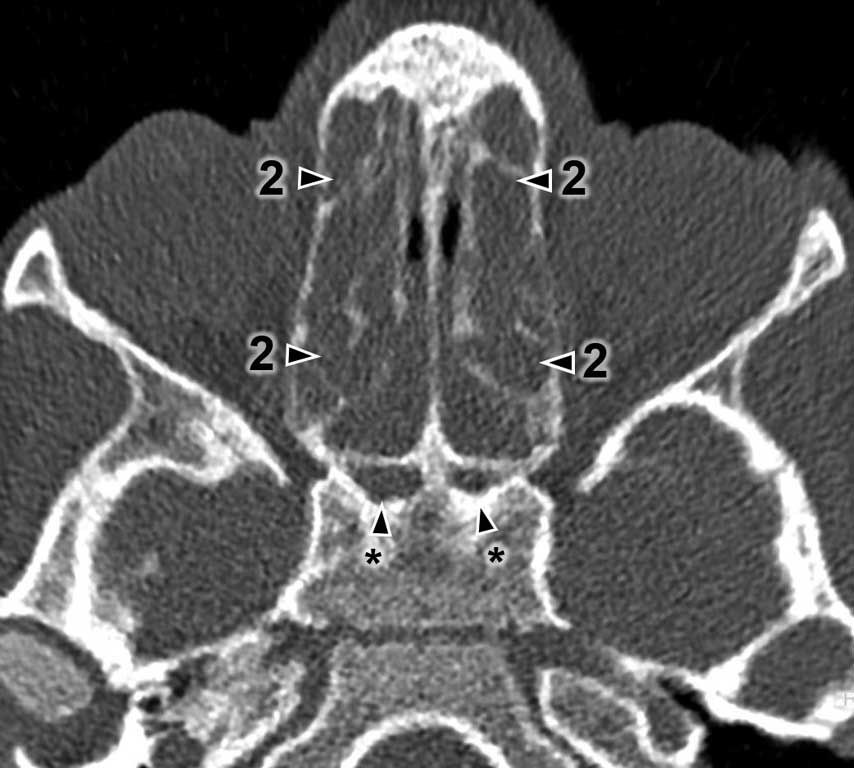
The CT results were assessed using the Lund-Mackay Score (LMS) and each examination was given from 0 to 24 scores. The detailed LMS scores are presented in table 1. Each sinus or group of sinuses was assigned from 0 to 2 scores, depending on the degree of shading. The completely shaded sinus in the CT image receives 2 scores, the sinus without mucosal oedema filled with air receives 0 scores. 1 score is given to the sinuses with any lesions that do not completely fill the lumen of the sinus. Moreover, 0 or 2 scores are assigned to the ostiomeatal complex if it is patent or obstructed. Figures 1 and 2 present a summary of CT images with the scores assigned. The asterisk means undeveloped sinuses. Images regard different patients.
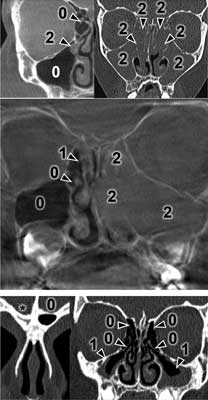
Fig. 1. Exemplary Modified Lund-Mackay scoring in different
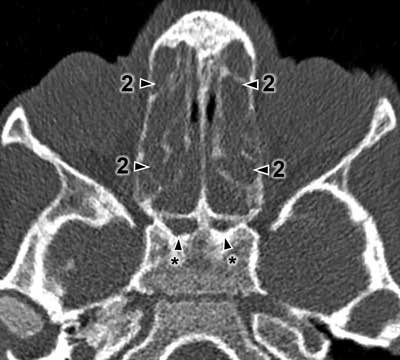
Fig. 2. Exemplary Modified Lund-Mackay scoring with undeveloped sphenoid sinuses
Tab. 1. Lund-Mackay Score
| Sinus | Right side | Left side |
| Maxillary | 0-1-2 | 0-1-2 |
| Frontal | 0-1-2 | 0-1-2 |
| Frontal ethmoid bone | 0-1-2 | 0-1-2 |
| Posterior ethmoid bone | 0-1-2 | 0-1-2 |
| Sphenoid | 0-1-2 | 0-1-2 |
| Ostiomeatal complex | 0-2 | 0-2 |
The same examinations were evaluated using the Modified Lund-Mackay Score (MLMS) which does not assign scores to undeveloped sinuses and ostiomeatal complex if it is subject to previous resection. The result is calculated proportionally to obtain a number between 0 and 24, according to the following formula:
where LMSmax means a maximum LMS result that might be obtained by the patient (24 reduced by 2 for each missing structure). Thus, the result between 0 and 24 is always obtained.
In the analysis of genotype of patients, the generally accepted division into mutation classes was taken into account. It was assumed that I, II and III class mutations are responsible for a more severe course of the disease, while IV, V and VI class mutations ? for milder one. This constitutes the basis for patients being divided into the groups with a “severe” and “mild” mutation (1, 6).
The links between the values were subject to a statistical analysis. In order to specify the correlation of linear parameters, the Pearson correlation coefficient was calculated, and the significance of the difference between LMS and MLMS in individual groups was set in the t-Student test. The results for which the p-coefficient was below 0.05 were considered to be statistically significant. Calculations were performed in MaxStat Lite, v. 3.60.
Results
34 patients aged from 3 to 17 years (median ? 10 years), 16 girls and 18 boys, were included in the study. One or more undeveloped paranasal sinuses were observed in 11 children (32%).
Severe mutation (of I-III classes) in both alleles was observed in 28 patients (82%), while 16 patients (47% of the entire group) were F508del homozygous. 6 patients (18%) presented mild mutation (of IV-VI classes) in at least one allele.
For the entire examined group, the median on LMS was 16 (range: 2-23), and on MLMS it equalled 17 (range: 2-24). The difference was statistically significant (p = 0.001). A significant difference between LMS and MLMS in the group of patients with severe and mild CFTR gene mutation was proved. LMS and MLMS medians were 17.5 and 18, respectively, in the group of patients with severe mutation and both for LMS and MLMS, they were 14.5 in the group of mild mutation (p = 0.044 and p = 0.031, respectively).
No significant correlation between age and LMS (Pearson coefficient 0.12 at p = 0.49) and between age and MLMS (Pearson coefficient 0.16 at p = 0.36) was demonstrated. No significant difference between LMS and MLMS depending on sex (p = 0.7 and p = 0.6, respectively) was also shown. LMS and MLMS in F508del homozygous patients did not differ significantly from the results in patients with a different allele set (p = 0.7 and p = 0.8, respectively).
Discussion
To reach diagnosis, in addition to identification of specific signs and symptoms, the applicable and valid European guidelines on the diagnosis and treatment of chronic sinusitis (EPOS 2012) require to obtain objective confirmation of the presence of a chronic inflammatory process within the nasal cavities and sinuses (7). For this purpose, CT scan of the sinuses is often made. The Lund-Mackay score is used to numerically determine and compare the severity of the inflammatory process within the sinuses (8). This scale was validated for adult patients with chronic sinusitis and it presents a good correlation with the severity of the inflammation process in sinuses (8).
Data in the literature defining possible links between the genotype of CF patients and the course of chronic sinusitis is limited and inconsistent. Most studies regarding chronic sinusitis in patients with cystic fibrosis are retrospective, and the study groups are small. There are relatively not many studies on children. Single reports prove a link between the genotype of F508del homozygous patients and more frequent diagnosis of chronic sinusitis (9) and more frequent qualification to Endoscopic Sinus Surgery (EES) (10) and occurrence of polyps (11), however other and more recent publications do not confirm these results (12). Patients with a severe mutation in both alleles of CFTR gene of I, II or III class more often suffer from, according to some researchers, nasal cavity polyps (13) and they present a higher score in the Lund-Mackay scale (14). In a recent study on a relatively large group of patients, Halderman et al. achieved a MLMS median of 13.88 in the group of patients with severe mutation in both alleles, and 8.06 in the group of patients with mild mutation in at least one allele (15). Similar results were reported by Ferril et al.: MLMS median for severe mutation in both alleles and mild mutation in at least one allele was 12.2 and 8.9, respectively (16). Abuzeid et al. however did not prove a difference in LMS results in adult patients with severe and mild mutation (17). The above data yet concerned adult patients with cystic fibrosis.
According to the authors, this is the first study that analyses the impact of cystic fibrosis genotype on the severity of sinus lesions in children. The results obtained are higher than in most studies regarding CF adult patients. The small study group and the retrospective observations are the limitations for this study. Children hospitalized in the Paediatric Otolaryn-gology Ward for various purposes were the group included in the study. Inflammation in the paranasal sinuses in these patients may be more evident than in the entire population of children with cystic fibrosis. Due to the relatively low occurrence of sinus-related symptoms, despite the radiological markers of severe chronic sinusitis in the CF patient population, these patients visit the otolaryngologist late. What is more, radiological tests with the use of ionizing radiation are less often instructed in children than in adults. According to the authors, these factors may lead to the selection of a group of more severely ill patients and the elevated results of LMS and MLMS in the examined population compared to the general population of children with CF. The results of LMS and MLMS obtained in this article are convergent with data from the literature. The LMS and MLMS results were 17 and 20 (18), 13.5 and 16 scores, respectively (19) in the studies comparing both scales in children with CF. Both of the above studies examined groups of children qualified for ESS, and they may therefore represent similar margin of error as the population in this study. The high percentage of paranasal sinus aplasia in patients with cystic fibrosis and the need for a modified Lund-Mackay score to assess CT in this group are also reflected in the literature (15, 18-22).
Conclusions
Lund-Mackay Score and Modified Lund-Mackay Score are used to assess the severity of sinus-related lesions in children with cystic fibrosis. A severe mutation in both alleles of the CFTR gene, of I-III classes, is associated with a greater progression of lesions shown in CR, while such a link was not proved for the genotype of F508del homozygous patients. The group of patients with a severe CFTR gene mutation presents higher scores for both LMS and MLMS. Due to possible sinus aplasia, the modified Lund-Mackay scale allows for error avoidance when comparing CT results of different patients.
Piśmiennictwo
1. Sands D, Pogorzelski A, Skoczylas-Ligocka A: Epidemiologia i organizacja opieki medycznej nad chorymi na mukowiscydozę w Polsce. [W:] Sands D (red.): Mukowiscydoza ? choroba wieloukładowa. Wyd. 1. Termedia, Poznań 2018: 15-24.
2. Illing EA, Woodworth BA: Management of the upper airway in cystic fibrosis. Curr Opin Pulm Med 2014; 20(6): 623-631.
3. Stephenson AL, Sykes J, Stanojevic S et al.: Survival Comparison of Patients With Cystic Fibrosis in Canada and the United States. Ann Intern Med 2017; 166: 537.
4. Walicka-Serzysko K, Skorupa W: Leczenie podtrzymujące przewlekłej choroby oskrzelowo-płucnej. [W:] Sands D (red.): Mukowiscydoza ? choroba wieloukładowa. Wyd. 1. Termedia, Poznań 2018: 147-154.
5. Zheng Z, Safi C, Gudis D: Surgical Management of Chronic Rhinosinusitis in Cystic Fibrosis. Medical Sciences 2019; 7: 57.
6. Welsh MJ, Smith AE: Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell 1993; 73: 1251-1254.
7. Fokkens WJ, Lund VJ, Mullol J et al.: EPOS 2012: European position paper on rhinosinusitis and nasal polyps 2012. A summary for otorhinolaryngologists. Rhinology Journal 2012; 50: 1-12.
8. Lund VJ, Kennedy DW: Staging for rhinosinusitis. Otolaryngol Head Neck Surg 1997; 117 (3 Pt 2): S35-40.
9. Jorissen MB, De Boeck K, Cuppens H: Genotype-Phenotype Correlations for the Paranasal Sinuses in Cystic Fibrosis. Am J Respir Crit Care Med 1999; 159: 1412-1416.
10. Becker SS, de Alarcon A, Bomeli SR et al.: Risk Factors for Recurrent Sinus Surgery in Cystic Fibrosis: Review of a Decade of Experience. Am J Rhinol 2007; 21: 478-482.
11. Sakano E, Ribeiro AF, Barth L et al.: Nasal and paranasal sinus endoscopy, computed tomography and microbiology of upper airways and the correlations with genotype and severity of cystic fibrosis. Int J Pediatr Otorhinolaryngol 2007; 71: 41-50.
12. Do BAJ, Lands LC, Saint-Martin C et al.: Effect of the F508del genotype on outcomes of endoscopic sinus surgery in children with cystic fibrosis. Int J Pediatr Otorhinolaryngol 2014; 78: 1133-1137.
13. Babinski D, Trawinska-Bartnicka M: Rhinosinusitis in cystic fibrosis: Not a simple story. Int J Pediatr Otorhinolaryngol 2008; 72: 619-624.
14. Berkhout MC, van Rooden CJ, Rijntjes E et al.: Sinonasal manifestations of cystic fibrosis: A correlation between genotype and phenotype? J Cyst Fibros 2014; 13: 442-448.
15. Halderman AA, Lee S, London NR et al.: Impact of high? versus low?risk genotype on sinonasal radiographic disease in cystic fibrosis. The Laryngoscope 2018; 129: 788-793.
16. Ferril GR, Nick JA, Getz AE et al.: Comparison of radiographic and clinical characteristics of low-risk and high-risk cystic fibrosis genotypes. Int Forum Allergy Rhinol 2014; 4: 915-920.
17. Abuzeid WM, Song C, Fastenberg JH et al.: Correlations between cystic fibrosis genotype and sinus disease severity in chronic rhinosinusitis. The Laryngoscope 2017; 128: 1752-1758.
18. Do BA, Lands LC, Mascarella MA et al.: Lund-Mackay and modified Lund-Mackay score for sinus surgery in children with cystic fibrosis. Int J Pediatr Otorhinolaryngol 2015; 79: 1341-1345.
19. Carter JM, Johnson BT, Patel A et al.: Lund-Mackay staging system in cystic fibrosis: a prognostic factor for revision surgery? Ochsner Journal 2014; 14(2): 184-187.
20. Orlandi RR, Wiggins RH III: Radiological Sinonasal Findings in Adults with Cystic Fibrosis. Am J Rhinol Allergy 2009; 23: 307-311.
21. Casserly P, Harrison M, O’Connell O et al.: Nasal endoscopy and paranasal sinus computerised tomography (CT) findings in an Irish cystic fibrosis adult patient group. Eur Arch Otorhinolaryngol 2014; 272: 3353-3359.
22. Manzini M, Schweiger C, Manica D et al.: Sinonasal computed tomography in pediatric cystic fibrosis: do we know the indications? Int J Pediatr Otorhinolaryngol 2018; 113: 204-207.