© Borgis - Medycyna Rodzinna 4/2008, s. 82-87
*Joanna Skulimowska, Jadwiga Sawecka
Diagnostyka molekularna hemofilii A
Molecular diagnostic of haemophilia A
Zakład Biochemii, Instytut Hematologii i Transfuzjologii w Warszawie
Kierownik Zakładu: dr Urszula Bany-Łaszewicz
Summary
Haemophilia A is a recessive, X-linked bleeding disorder affecting approximately 1/5000 males due to a deficiency or dysfunction of blood coagulation factor VIII. Approximately one third of cases have no prior history of haemophilia A (sporadic haemophilia). The aim of this study is to present a diagnostical strategy for carriership determination and prenatal diagnostics of haemophilia A. The severity of haemophilia A should be determined first because it influences choice of diagnostic strategy. A wide range of different mutation types has been identified, which occur along the entire length of the factor VIII gene. In 20% of haemophilia A cases an inversion in intron 22 of FVIII is the causative mutation, which always leads to severe disease. The second most common mutation in severe disease is the intron 1 inversion mutation, which occurs with a frequency 1-5%. Intron 22 inversion in FVIII gene was directly examined by Southern blot analysis, intron 1 inversion was identified by dual PCR assay. If these mutations are absent, further analysis includes DNA sequencing of those regions in which a mutation is suspected. We used SSCP and HD as screening methods for detecting abnormal fragment. Families with mild and moderate haemophilia A were testing directly by linkage analysis or sequencing. When family has not identified a causative mutation, linkage analysis employing DNA polymorphic markers was used. It is essential to have heterozygous marker in the mother to carry out efficient disease gene tracking. We used dinucleotide polymorphisms in intron 1, 13, 22 and 24.

Wstęp
Hemofilia A jest jedną z najczęściej występujących skaz krwotocznych, niezależną od pochodzenia geograficznego i etnicznego chorych. Hemofilia A charakteryzuje się wrodzonym brakiem, niedoborem lub nieprawidłową strukturą VIII czynnika krzepnięcia. Aktywny czynnik VIII (VIIIa) wraz z aktywnym czynnikiem IX, jonami wapnia i fosfolipidami tworzy kompleks zwany tenazą, którego zadaniem jest aktywacja czynnika X (1). Zaburzenia w kaskadzie krzepnięcia powodują skłonność do nadmiernych krwawień, samoistnych lub na skutek niewielkich urazów. Powtarzające się epizody krwawień następują zwykle do stawów, mięśni, przewodu pokarmowego, mogą występować wylewy podskórne, krwiomocz. Poważnymi późnymi powikłaniami choroby są artropatia hemofilowa oraz zakażenia wirusowe. Nasilenie skazy krwotocznej jest uzależnione od stopnia niedoboru czynnika VIII. Wyróżniamy postać ciężką, w której poziom czynnika VIII nie przekracza 1% normy, umiarkowaną, z czynnikiem VIII na poziomie 1-5% oraz łagodną, powyżej 5% (2). Diagnostyka hemofilii A opiera się na stwierdzeniu wydłużonego czasu kaolinowo-kefalinowego (APTT) oraz obniżeniu aktywności FVIII: C przy prawidłowych: liczbie płytek krwi, czasie krwawienia, czasie protrombinowym. Leczenie chorych to przede wszystkim leczenie substytucyjne, polegające na uzupełnianiu niedoboru czynnika VIII. Problemem terapeutycznym jest pojawienie się, najczęściej we wczesnym dzieciństwie, krążącego antykoagulantu (inhibitora) wobec czynnika VIII. Stężenie przeciwciał wyraża się w jednostkach Bethesda w mililitrze osocza chorego (j.B./ml). Chorzy, u których miano nie przekracza 5 j.B., zaliczani są do grupy LR, słabo reagującej na bodziec antygenowy (low responders). Wzrost miana inhibitora powyżej 5 j.B. lokuje chorych w grupie HR (high responders) (3). Eliminację inhibitora, skuteczną w większości przypadków, uzyskuje się poprzez indukowanie tolerancji immunologicznej wobec VIII czynnika krzepnięcia.
Od 1991 r. w Instytucie Hematologii i Transfuzjologii prowadzony jest ogólnopolski komputerowy rejestr chorych na hemofilię i pokrewne zaburzenia krzepnięcia krwi, obejmujący obecnie około 2000 pacjentów z hemofilią A. W populacji polskiej stanowi ona ponad 60% wszystkich przypadków wrodzonych zaburzeń krzepnięcia krwi. Występowanie choroby szacuje się z częstością 1:12 300 mieszkańców (1:5600 mężczyzn). W Polsce postać ciężka choroby stanowi blisko 60% zarejestrowanych przypadków (4). Hemofilia A jest dziedziczona w sposób recesywny związany z płcią, kobiety są nosicielkami a chorują mężczyźni, choć znane są bardzo rzadkie przypadki chorych kobiet. Wszystkie córki chorego są nosicielkami, a wszyscy synowie są zdrowi. Matka nosicielka przekazuje defektywny gen z 50% prawdopodobieństwem (ryc. 1).
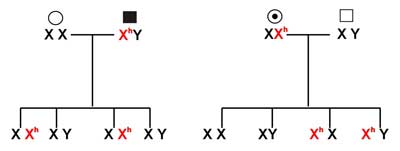
Ryc. 1. Schemat dziedziczenia hemofilii A.
Budowa genu
Gen kodujący czynnik VIII jest zlokalizowany w części dystalnej długiego ramienia chromosomu X, w lokus Xq28 (ryc. 2). Gen czynnika VIII jest jednym z większych genów ludzkich, obejmuje 186kb i składa się z 26 eksonów, przedzielonych intronami. Został wyizolowany, sklonowany i scharakteryzowany w 1984 roku (5). FVIII mRNA o wielkości około 9 kb zawiera nukleotydy kodujące peptyd sygnałowy i dojrzały peptyd, składający się z 2332 aminokwasów. Białko to, tzw. globulina antyhemofilowa składa się z 6 podjednostek i dwóch łańcuchów: lekkiego (od strony C-końca, 80 kDa, A1-A2-B) i ciężkiego (200 kDa, A3-C1-C2) (6, 7). Jest syntetyzowany głównie przez wątrobę, w mniejszym stopniu przez komórki śledziony i węzłów chłonnych. Czynnik VIII krąży jako nieaktywowany pro-kofaktor w kompleksie z czynnikiem von Willebranda.
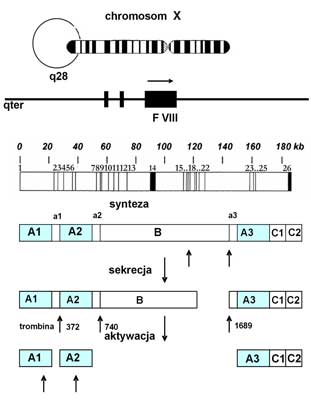
Ryc. 2. Budowa genu czynnika VIII.
Diagnostyka metodami biologii molekularnej
Od czasu sklonowania genu czynnika VIII rozpoczął się gwałtowny rozwój badań nad hemofilią A, opartych na metodach biologii molekularnej. Wdrożono diagnostykę molekularną choroby, obejmującą trzy zasadnicze cele:
1. diagnostyka nosicielstwa,
2. analiza prenatalna,
3. identyfikacja indywidualnych mutacji sprawczych.
Realizacja tych celów opiera się na dwóch strategiach:
1. analiza sprzężeń genetycznych,
2. bezpośrednie wykrywanie mutacji.
Badania wykonywane w Pracowni Biologii Molekularnej Zakładu Biochemii Instytutu Hematologii i Transfuzjologii obejmują zarówno diagnostykę nosicielstwa, analizę prenatalną, jak też identyfikację indywidualnych mutacji sprawczych.
Materiał i metody
Genomowy DNA izolowano z pełnej krwi obwodowej metodą z zastosowaniem 6M NaCL do odbiałczania preparatów DNA. Materiałem do badań prenatalnych były kosmki trofoblastu. Analizę polimorfizmów prowadzono z zastosowaniem techniki PCR oraz elektroforezy w natywnym żelu poliakrylamidowym, barwionym srebrem. Obecność mutacji inwersyjnej intronu 22 oznaczano posługując się metodą Southern blotting i sondą sklonowaną w plazmidzie p482.6, wyznakowaną (α32P) dCTP, allele identyfikowano autoradiograficznie. Mutację inwersyjną intronu 1 badano za pomocą multipleks PCR i elektroforezy. Reakcję sekwencjonowania prowadzono metodą cykliczną z użyciem polimerazy DNA Ampli Taq, FS i terminatorów znakowanych barwnikami fluorescencyjnymi, pochodnymi dichloroaminy (A-dR6G, C-dROX, G-R110, T-dTAMRA). Elektroforezę prowadzono w warunkach denaturujących na 4% żelu poliakryloamidowym w sekwenatorze ABI PRISM 377.
Analiza sprzężeń genetycznych
Złożoność genu czynnika VIII oraz heterogenna natura wykrywanych mutacji powoduje trudności w bezpośredniej detekcji mutacji sprawczej u chorego i jego krewnych. Zatem w wielu laboratoriach wykorzystuje się metodę pośrednią, służącą do stwierdzenia stanu nosicielstwa w rodzinie z nieznaną mutacją, czyli diagnostykę poprzez analizę sprzężeń genetycznych (linkage analysis) (8). Wybrane markery znajdują się wewnątrz genu, zazwyczaj w jego niekodujących częściach. Stosuje się markery będące polimorfizmami restrykcyjnymi (RFLP, restriction fragment length polymorphism, polimorfizm długości fragmentów restrykcyjnych), a częściej polimorfizmami mikrosatelitarnymi, charakteryzującymi się zmienną osobniczo liczbą krótkich, kilkunukleotydowych sekwencji (STR – short tandem repeats, krótkie tandemowe powtórzenia). Powtórzenia krótkich motywów sekwencji DNA są rozproszone w całym genomie człowieka. Najliczniejsze z nich, dwunukleotydowe powtórzenia cytozyny i adeniny (CA), występują średnio co kilka tysięcy nukleotydów. Metoda umożliwia śledzenie przekazywania polimorficznych alleli oraz zmutowanego allelu w obrębie rodziny obarczonej chorobą (ryc. 3). Badania polimorfizmu mają pewne ograniczenia: wysoka liczba mutacji de novo (w polskim rejestrze w około 50% przypadków wywiady w kierunku występowania hemofilii A u innych członków rodziny były ujemne), dostępność wszystkich niezbędnych członków rodziny. W przypadku hemofilii sporadycznej można jedynie wykluczyć stan nosicielstwa, gdy badana nie dziedziczy allelu sprzężonego z chorobą. Homozygotyczność nosicielki uniemożliwia identyfikację i śledzenie dziedziczenia w rodzinie nieprawidłowego genu, zatem najbardziej przydatne są wieloalleliczne układy STR.
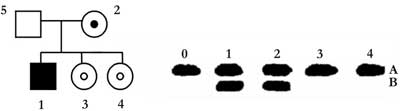
Ryc. 3. Przykładowa analiza polimorfizmu intronu 22 w rodzinie W.
Stosowane przez nas metody obejmują analizę polimorfizmu:
1. polimorfizm dwunukleotydowych powtórzeń w intronie 13 genu czynnika VIII (9, 10),
2. polimorfizm dwunukleotydowych powtórzeń w intronie 22 genu czynnika VIII (9, 10),
3. polimorfizm dwunukleotydowych powtórzeń w intronie 24 genu czynnika VIII (11),
4. polimorfizm dwunukleotydowych powtórzeń w intronie 1 genu czynnika VIII (12),
5. dimorfizm miejsca restrykcyjnego BclI w intronie 18 genu czynnika VIII (13).
Celem badania jest ustalenie stanu nosicielstwa u kobiet [3] i [4], sióstr chorego [1], córek pewnej nosicielki [2].
U chorego [1] występuje allel oznaczony umownie literą [A], sprzężony z hemofilią. Matka-nosicielka [2]-heterozygota, posiada allel sprzężony z chorobą [A] oraz allel sprzężony z prawidłową postacią genu czynnika VIII, oznaczony jako [B].
Córka [3] odziedziczyła od ojca allel [A], zatem od matki allel [B], odpowiadający prawidłowemu genowi, nie jest więc nosicielką. Córka [4] odziedziczyła od matki allel [A], sprzężony z chorobą, czyli jest nosicielką. Jednocześnie jest homozygotą, toteż polimorfizm intronu 22 nie będzie informatywny w ewentualnej diagnostyce jej potomstwa.
Bezpośrednie wykrywanie mutacji
Mutacje sprawcze w hemofilii A rozproszone są wewnątrz całego genu. Liczba opisanych mutacji wzrasta z roku na rok i wg ostatnich danych przekroczyła 1200. W genie czynnika VIII występują praktycznie wszystkie rodzaje mutacji począwszy od rearanżacji, delecji poprzez insercje i duplikacje do mutacji punktowych (zmiany sensu i nonsensownych) i splicingowych (14). Diagnostyka wymaga zatem indywidualnego podejścia w badaniu każdej rodziny. Wyjątkiem jest tzw. mutacja inwersyjna intronu 22, która występuje u niemal 50% pacjentów z ciężką postacią choroby (15, 16). Postać choroby jest więc bardzo istotną informacją, od niej bowiem zależy wybór strategii postępowania diagnostycznego. W przypadku hemofilii ciężkiej pierwszym etapem jest badanie obecności mutacji inwersyjnych, najpierw intronu 22, następnie intronu 1. Umiarkowana i łagodna postać choroby wywołana jest głównie mutacjami punktowymi. Wykrywanie ich rozpoczyna amplifikacja wszystkich eksonów genu wraz z obszarami flankującymi, a następnie wykorzystuje się techniki przesiewowe albo bezpośrednie sekwencjonowanie.
Mutacja inwersyjna intronu 22
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Radziwon P, Kłoczko J, Kiss B: Współczesna teoria aktywacji i kontroli krzepnięcia krwi. Przew Lek 2004; 11: 50-56. 2. Pandey GS, Mittal B.: Molecular diagnosis in hemophilia A. J Postgrad Med 2001; 47: 274-80. 3. Lillicrap D: The role of immunomodulation in the management of Factor VIII inhibitors. Hematology Am Soc Hematol Educ Program 2006; 421-5. 4. Windyga J et al.: Hemofilia i pokrewne skazy krwotoczne w Polsce. Pol Arch Med 2004; 112: 1197-202. 5. Gitschier J et al.: Characterization of the human factor VIII gene. Nature 1984; 312: 326-30. 6. Shen BW et al.: The tertiary structure and domain organization of coagulation factor VIII. Blood 2008; 111: 1240-7. 7. Fang H, Wang L, Wang H: The protein structure and effect of factor VIII. Thromb Res 2007; 119: 1-13. 8. Peyvandi F: Carrier detection and prenatal diagnosis of hemophilia in developing countries. Semin Thromb Hemost 2005; 31: 544-54. 9. Lalloz MRA et al.: Hemophilia A diagnosis by simultaneous analysis of two variable dinulceotide tandemrepeats within the factor VIII gene. Br J Haematol 1994; 86: 804-9. 10. Sawecka J, Skulimowska J, Kościelak J: Zastosowanie uproszczonej metody analizy polimorfizmu dwunukleotydowych powtórzeń w intronach 13 i 22 genu czynnika VIII do wykrywania nosicielstwa hemofilii A. Acta Hematol Pol 1997; 28: 267-271. 11. Kim J-W et al.: Identification of new dinucleotide-repeat polymorphisms in the factor VIII gene using fluorescent PCR. Hemophilia 2005; 11: 38-42. 12. Tizzano E et al.: Utility of a (GT)n dinucleotide repeat In intron 1 of the factor 8 gene for haemophilia A carrier diagnosis. Haemophilia 2005; 11: 142-4. 13. Kogan SC, Doherty M, Gitschier J: An improved method for prenatal diagnosis of genetic diseases by analysis of amplified DNA sequencies. Application to Hemophilia A. The New England Journal of Medicine 1987; 317: 985-990. 14. Haemophilia A Mutation Database (http://europium.csc.mrc.ac.uk). 15. Lakich D et al.: Inversions disrupting the factor VIII gene are a common cause for severe haemophilia A. Nat Genet 1993; 5: 236-41. 16. Naylor J et al.: Characteristic mRNA abnormality found in half the patients with severe haemophilia A is due to large DNA inversions. Hum Mol Genet 1993; 2: 1773-8. 17. Bagnall RD, Giannelli F, Green PM: Int22h-related inversions causing hemophilia A: a novel insight into their origin and a new more discriminant PCR test for their detection. J Thromb Haemost 2006; 4: 591-8. 18. Sawecka J et al.: Prevalence of the intron 22 inversion of the factor VIII gene and inhibitor development in Polish patients with severe hemophilia A Arch. Immunol. Ther Exp 2005; 53: 352-6. 19. Antonarakis SE et al.: Factor VIII gene inversions in severe hemophilia A: Results of an international consortium study. Blood 1995; 86: 2206-12. 20. Bagnall RD et al.: Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood 2002; 99: 168-74. 21. Sawecka J et al.: Inwersja intronu 1 genu cz.VIII u pacjentów chorych na ciężką hemofilię A. Acta Haematol Pol 2006; 37: 61-5. 22. Keeney S, Mitchell M, Goodeve A: The molecular analysis of haemophilia A: a guideline from the UK haemophilia centre doctors´ organization haemophilia genetics laboratory network. Haemophilia 2005; 11: 387-97. 23. Ludlam CA et al.: A framework for genetic service provision for haemophilia and other inherited bleeding disorders Haemophilia. 2005; 11: 145-63. 24. David D et al.: Analysis of the consequences of premature termination codonc within factor VIII coding sequences. J Thromb Haemost 2003; 1: 139-46.