© Borgis - Postępy Nauk Medycznych 7-8/2007, s. 333-337
*Barbara Nasiłowska-Adamska1, Iwona Malinowska2
Znaczenie mutacji genu FLT 3 u chorych na ostre białaczki – praca poglądowa
The role of FLT3gene mutations in acute leukemia – review
1Klinika Transplantacji Komórek Krwiotwórczych Instytutu Hematologii i Transfuzjologii w Warszawie
Kierownik Kliniki: prof. dr hab. med. Bożena Mariańska
2Katedra i Klinika Pediatrii, Hematologii i Onkologii AM w Warszawie
Kierownik Kliniki: prof. dr hab. n. med. Michał Matysiak
Streszczenie
FLT3 jest receptorem posiadającym aktywność kinazy tyrozynowej (KT) i niezbędnym we wczesnych etapach hematopoezy. Przyłączenie liganda FLT3do receptora FLT3indukuje jego aktywność kinazową. W komórkach białaczkowych obserwowana jest stała aktywacja receptora w wyniku autokrynnego lub parakrynnego wydzielania liganda FLT3, mutacji aktywujących lub nadekspresji FLT3.
Najczęściej występujące typy mutacji genu kodującego receptor FLT3to: wewnętrzna tandemowa duplikacja (ITD, internal tandem duplication) oraz mutacje punktowe występujące w domenie kinazowej.
Liczne badania wskazują na niekorzystne prognostycznie znaczenie mutacji FLT3-ITD u chorych na ostre białaczki. Nadekspresję genu FLT3stwierdza się u 50% chorych na ostre białaczki, mutację FLT3-ITD u 15-30%. Defekt receptora jest przyczyną nieprawidłowego przekazywania sygnałów wewnątrzkomórkowych, prowadzącego do niekontrolowanej proliferacji komórek białaczkowych. Stwierdzenie mutacji FLT3-ITD jest związane z gorszym rokowaniem i jest wskazaniem do intensyfikacji leczenia, zastosowania jednego z nowych inhibitorów FLT3oraz kwalifikacji do szybkiego wykonania transplantacji komórek krwiotwórczych.
Summary
FLT3 is a tyrosine kinase receptor (TK) necessary in the early phases of hematopoiesis. FLT3ligand binds to FLT3receptor and induces activation of TK. In leukemic cells, FLT3receptor is constitutively activated through autocrine or paracrine secretion of the FLT3ligand, activating mutations or overexpression of FLT3.
The most common types of FLT3mutations are: internal tandem duplication (ITD) of juxtamembrane domain and the point mutations in kinase domain. Many studies presented the FLT3-ITD mutation as a marker of a poor prognosis in the patients with leukemia. Overexpression of FLT3gene and FLT3-ITD has been found, respectively in 50% and 15-30% patients with acute leukemias.
Defects of FLT3receptor lead to abnormal signal transduction and uncontrolled proliferation of leukemic cells. Patients with FLT3-ITD require intensification of therapy, application of FLT3inhibitors and earlier transplantation of hematopoietic stem cells.

FLT3
FLT3 [fms-like tyrosine kinase 3; fetal liver tyrosine kinase 3 lub stem cell tyrosine kinase 1 (STK1)] należy do III klasy receptorowych kinaz tyrozynowych (KT). Schemat receptora przedstawiono na rycinie 1 (1). Gen kodujący receptor FLT3jest zlokalizowany na chromosomie 13 i składa się z 24 egzonów (2). Ekspresję genu FLT3stwierdza się w prawidłowych, progenitorowych komórkach hematopoetycznych szpiku oraz limfopoetycznych grasicy i węzłów chłonnych (2). W warunkach prawidłowych interakcja receptora FLT3z ligandem FLT3 (FL) indukuje aktywność KT i w ten sposób inicjuje przekazywanie sygnałów wewnątrzkomórkowych regulujących proliferację i różnicowanie komórek (1-4). W procesie różnicowania komórek krwiotwórczych ekspresja FLT3ulega osłabieniu i zanika (1) .
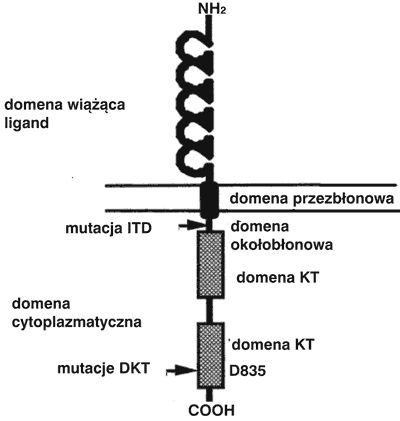
Ryc. 1. Struktura FLT3i lokalizacja mutacji. Opis w tekście.
Mutacje FLT3w ostrych białaczkach
W komórkach białaczkowych obserwuje się zwiększoną aktywność KT receptora FLT3w wyniku:
1. nadekspresji FLT3,
2. autokrynnego i parakrynnego wydzielania FL,
3. mutacji aktywujących.
Nadekspresję FLT3, ocenianą w ilościowych badaniach łańcuchowej reakcji polimerazy i immunofenotypowaniu (CD135), stwierdza się w komórkach białaczkowych u 50% chorych na ostrą białaczkę nielimfoblastyczną (OBNL) i limfoblastyczną (OBL) z linii B-komórkowej oraz w części przypadków OBL T-komórkowej i w przełomie blastycznym przewlekłej białaczki szpikowej (PBSz) (1).
Wykazano również zwiększoną ekspresję FLT3u 5-20% dzieci chorych na OBL z obecnością częściowej tandemowej duplikacji (PTD, partial tandem duplication) genu MLL (mixed lineage leukemia) lub hiperdiploidalną liczbą chromosomów (powyżej 50) (5). Autokrynną i parakrynną aktywację receptora FLT3stwierdzono w komórkach białaczkowych chorych na OBNL i OBL z linii B-komórkowej (6).
Najczęściej występujące typy mutacji genu kodującego receptor FLT3to: wewnętrzna tandemowa duplikacja (ITD, internal tandem duplication), która występuje we fragmencie genu kodującym domenę okołobłonową receptora oraz mutacje punktowe występujące w drugiej domenie kinazowej (DKT) (ryc. 1). Defekt receptora jest przyczyną stałego przekazywania sygnału z receptora do białek efektorowych i prowadzi do niekontrolowanej proliferacji komórek białaczkowych. Mutacje genu kodującego receptor FLT3i powodujące jego aktywację odgrywają istotną rolę w procesie leukemogenezy.
Należy zaznaczyć, że oba typy mutacji wywołują stałą, niezależną od liganda aktywację receptora FLT3 (7). Mutacja ITD zaburza funkcję regulatorową obszaru okołobłonowego receptora a mutacja punktowa DKT zaburza funkcję pętli aktywującej receptora (7).
Mutacja FLT3-ITD jest jedną z najczęściej występujących mutacji somatycznych i jest wykrywana u około 15-30% chorych na OBNL (1). Zaobserwowano, że nadekspresja FLT3oraz mutacja ITD występują istotnie częściej w obecności mutacji PTD genu MLL (8, 9, 10). Mutacje DKT (D835) domeny kinazowej są wykrywane u 8-12% chorych na OBNL (11).
Prowadzone są bardzo intensywne badania nad znaczeniem mutacji FLT3w chorobach nowotworowych układu krwiotwórczego.
Trzy badania obejmujące duże grupy chorych na OBNL wykazały niekorzystny wpływ występowania FLT3-ITD na czas wolny od wydarzeń (EFS, event free survival) z powodu dużego odsetka nawrotów w tej grupie chorych oraz na czas całkowitego przeżycia (OS, overall survival) (12, 13, 14). Schnittger i wsp. przeprowadzili badanie kliniczne w grupie 1003 chorych na OBNL i stwierdzili występowanie mutacji ITD u 23% chorych z istotnie krótszym EFS: 7,4 m-ce vs. 12,6 m-cy (12). Natomiast Kottaridis i wsp. w grupie 854 chorych na OBNL stwierdzili mutację FLT3-ITD u 27% chorych i istotnie krótszy 5-letni DFS (30% vs. 46%), EFS (23% vs. 39%) i OS (32% vs.44%) (13). W badaniach tych występowanie mutacji ITD było związane z leukocytozą, dużym odsetkiem blastów w szpiku, brakiem aberracji cytogenetycznych poza obecnością t(15;17). Natomiast ITD współwystępuje rzadko w OBNL z t(8;21), inv(16), t(16;16) oraz z aberracjami 11q23 i złożonymi aberracjami cytogenetycznymi.
Podkreślono również bardzo niekorzystne znaczenie całkowitej lub częściowej utraty niezmutowanego tzw. „dzikiego typu” FLT3u chorych z mutacją ITD (14).
Częste występowanie mutacji ITD zaobserwowano w OBNL M3 (wg klasyfikacji FAB), a w szczególności podtypie M3v. Mutacje w tym typie białaczki są przyczyną szczególnie niekorzystnego przebiegu choroby, z małym odsetkiem uzyskiwanych remisji, częstymi nawrotami i krótkim czasem przeżycia chorych (15, 16, 17).
U osób powyżej 60 lat nie wykazano znaczenia prognostycznego mutacji FLT3-ITD, co prawdopodobnie wynika z wyjątkowo złej prognozy w tej grupie wiekowej.
U dzieci chorych na OBNL do 10 roku życia mutacje ITD występują wyjątkowo rzadko a w całej populacji dziecięcej dotyczą 15% chorych (1). Meshinchi i wsp. wykazali jednak, że EFS u dzieci bez FLT3-ITD wynosiło 44% a w grupie z obecnością tej mutacji tylko 7% (18).
Częstość mutacji DKT u dzieci chorych na OBNL jest zbliżona do częstości jej występowania u dorosłych. Należy jednak zauważyć, że większość badań wskazuje na brak znaczenia prognostycznego mutacji punktowych DKT i bardziej pomyślny przebieg choroby w przypadku jej obecności w porównaniu do FLT3-ITD (1, 12). Może to być wynikiem różnic w mechanizmach przekazywania sygnałów komórkowych do białek efektorowych.
U części chorych nie stwierdza się mutacji FLT3w chwili rozpoznania OBNL, ale aberracja ta może pojawić się we wznowie białaczki (12). U 84% chorych na OBNL FLT3-ITD jest stwierdzana w momencie rozpoznania choroby i w przypadku nawrotu (19). U pozostałych 16% chorych nie ujawnia się ponownie w komórkach białaczkowych w nawrocie choroby. Natomiast mutacja DKT pojawia się u ponad 50% chorych w nawrocie OBNL (20).
Mutacje FLT3-ITD występują znacznie rzadziej u chorych na OBL (<1%) i zwykle dotyczą przypadków białaczki dwufenotypowej (1).
Mutację FLT3stwierdzono również u 3-5% chorych z zespołami mielodysplastycznymi (MDS). U pacjentów z MDS bez mutacji FLT3w chwili diagnozy aberracja ta może się pojawić w okresie transformacji do ostrej białaczki (21).
Należy wspomnieć, że wywołana mutacją aktywacja receptora FLT3równocześnie powoduje pośrednią lub bezpośrednią fosforylację i aktywację innych białek efektorowych: PI-3-ki/AKT, RAS/MAPK i JAK/STAT5 i reguluje wiele procesów wewnątrzkomórkowych (1, 2).
Za niekorzystny przebieg choroby i oporność na leczenie może odpowiadać kilka mechanizmów. Zakwalifikowanie chorego do odpowiedniej grupy prognostycznej powinno uwzględniać opisane czynniki ryzyka, m.in. ekspresję genu mdr1 (multidrug resistance gene 1) i ilość glikoproteiny P (PgP) (22, 23). Marzac i wsp. udowodnili, że FLT3-ITD i PgP są niezależnymi oraz addycyjnymi niekorzystnymi czynnikami rokowniczymi u chorych na OBNL (23). Odpowiednia stratyfikacja chorych do grup prognostycznych wpływa na wybór właściwej terapii i poprawę wyników leczenia, głównie u chorych na OBNL z prawidłowym kariotypem należących do grupy pośredniego ryzyka cytogenetycznego.
Stwierdzenie mutacji FLT3pogarsza rokowanie i jest wskazaniem do intensyfikacji leczenia, pozwala zastosować jeden z nowych inhibitorów receptora FLT3oraz kwalifikuje do szybkiego wykonania transplantacji komórek krwiotwórczych (SCT, stem cell transplantation). Z drugiej strony pojawiają się kontrowersje dotyczące kwalifikacji pacjentów z mutacją FLT3do SCT (24, 25). Gale i wsp. przeprowadzili badanie w grupie 1135 chorych na OBNL oceniające wyniki transplantacji w zależności od obecności mutacji FLT3i wywnioskowali, że transplantacja nieistotnie statystycznie zmniejsza ryzyko nawrotu choroby oraz skraca czas EFS i OS u chorych z mutacją FLT3-ITD (25). Natomiast grupa niemiecka wykazała, że auto- lub allo-SCT u pacjentów z FLT3-ITD (31,5% z 999 chorych) istotnie zmniejsza ryzyko nawrotu białaczki (59% vs. 94%) i wydłuża OS (4,5 lata: 46% vs. 21%) w porównaniu do pacjentów nie poddanych SCT (26).
Powyższe wyniki sugerują brak jednoznacznie ustalonych wskazań do SCT u chorych z mutacją FLT3-ITD. Niezbędne jest wykonanie dalszych badań w dużych grupach chorych w celu poznania wpływu mutacji FLT3na wyniki SCT.
Badania przedkliniczne inhibitorów receptora FLT3
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Small D.: FLT3 mutations: Biology and treatment. Hematology 2006; 178-184.
2. Kottaridis P.D., et al.: FLT3 mutations and leukemia-review. Br. J. Haematology 2003; 122: 523-538.
3. Gilliland O.G., Griffin J.O.: The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002; 100: 1532-1542.
4. Stirewalt D.L., Radich J.P.: The role of FLT3 in hematopoietic malignancies. Nat. Rev. Cancer., 2003; 3: 650-665.
5. Armstrong S.A., et al.: FLT3 mutations in childhood acute lymphoblastic leukemia. Blood. 2004; 1 03: 3544-3546.
6. Zheng R., et al.: FLT3 ligand causes autocrine signaling in acute myeloid leukemia cells. Blood. 2004; 103: 267-274.
7. Griffith J., et al.: The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol. Cell., 2004; 13: 169-178.
8. Libura M., et al.: FLT3 and MLL intragenic abnormalities in AML reflect a common category of genotoxic stress. Blood 2003; 102: 2198-2204.
9. Ozeki K., et al.: Biologic and clinical significance of the FLT3 transcript level In acute myeloid leukemia. Blood 2004; 103: 1901-1908.
10. Stendel C., et al.: Comparative analysis of MLL partial tandem duplication and FLT3 internal tandem duplication mutations in 956 adult patients with AML. Genes Chromosomes Cancer 2003; 37: 237-51.
11. Yamamoto Y., et al.: Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001; 97: 2434-2439.
12. Schnittger S., et al.: Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype and prognosis in the AMLCG study and usefullness as a marker for the detection of minimal residual disease. Blood 2002; 100: 59-66.
13. Kottaridis D.P., et al.: The presence of a FLT3 tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from United Kingdom Medical Council AML 10 and 12 trials. Blood 2001; 98: 1752-1761.
14. Thiede C., et al.: Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002; 99: 4326-35.
15. Hasan S.K., et al.: Impast of FLT3 internal tandem duplication on Indian acute promyelocytic leukemia patients: prognostic implications. Hematology 2007; 12: 99-101.
16. Gale R.E., et al.: The relationship between FLT3 mutations status, biological characteristics and response to targeted therapy in acute promyelocytic leukemia. Blood 2005; 106: 3768-76.
17. Callens C., et al.: Prognostic implication of FLT3 and Ras gene mutations in patients with acute promyelocytic leukemia (APL): a retrospective study from the European APL Group. Leukemia 2005; 19: 1153-60.
18. Meshinehi S., et al.: Prevalence and prognostic significance of Flt3 internal tandem duplication in pediatric acute myeloid leukemia. Blood. 2001; 97: 89-94.
19. Shih L.V., et al.: Internal tandem duplication of FLT3 in relapsed acute myeloid leukemia: a comparative analysis of bone marrow samples from 108 adult patients at diagnosis and relapse. Blood. 2002; 100: 2387-2392.
20. Shih L.V., et al.: Heterogeneous patterns of FLT3 Asp(835) mutations in relapsed de novo acute myeloid leukemia: a comparative analysis of 120 paired diagnostic and relapse bone marrow samples. Clin. Cancer Res., 2004; 10: 1326-1332.
21. Shih L.V., et al.: Acquisition of FLT3 or N-ras mutations is frequently associated with progression of myelodysplastic syndrome to acute myeloid leukemia. Leukemia. 2004; 18: 466-475.
22. Baldus C.D., et al.: Clinical outcome of de novo acute myeloid leukemia patients with normal cytogenetics is affected by molecular genetic alterations: a concise review. Br. J. Haematology 2007; 137: 387-400.
23. Marzac C., et al.: Flt3 internal tandem duplication and P-glycoprotein functionality in 171 patients with acute myeloid leukemia. Clin. Cancer Res., 2006; 12: 7018-24.
24. Doubek M., et al.: Is FLT3 internal tandem duplication significant indication for allogeneic transplantation in acute myeloid leukemia? An analysis of patients from the Czech acute Leukemia Clinical Register (ALERT). Neoplasma 2007; 54: 89-95.
25. Gale R.E., et al.: No evidence that FLT3 status should be considered as an indicator for transplantation in acute myeloid leukemia (AML): an analysis of 1135 patients, excluding acute promyelocytic leukemia, from the UK MRC AML 10 and 12 trials. Blood 2005; 106: 3658-3665.
26. Schlenk R.F., et al.: Postremission therapy with an allogeneic transplantation from an HLA-matched family donor seems to overcome the negative prognostic impact of FLT3-ITD in younger patients with acute myeloid leukemia exhibiting a normal karyotype (abstract). Blood 2005; 106: 662a.
27. Levis M., et al.: A FLT3 tyrosine kinase inhibitor is selectively cytotoxic to acute myeloid leukemia blasts harboring FLT3 internal tandem duplication mutations. Blood. 2001; 98: 885-887.
28. Shankar D.B., et al.: ABT-869 a multi-targeted receptor tyrosine kinase inhibitor: inhibition of FLT3 phosphorylation and signaling in acute myeloid leukemia. Blood 2007 (Epub ahead of print).
29. Lierman E., et al.: The ability of sorafenib to inhibit oncogenic PDGFRbeta and FLT3 mutants and overcome resistance to Rother small molekule inhibitors. Haematologica 2007; 92: 27-34.
30. Auclair D., et al.: Antitumot activity of sorafenib in FLT3-driven leukemic cells. Leukemia 2007; 21: 439-45.
31. Chow L.Q, Eckhardt S.G.: Sunitinib: from rational design to clinical efficacy. J. Clin. Oncol., 2007; 25: 884-96.
32. Griessinger E., et al.: AS602868, a dual inhibitor of IKK2 and FLT3 to target AML cells. Leukemia 2007 (Epub ahead of print).
33. Levis M., Smali O.: FLT3 tyrosine kinase inhibitors. Int. J. Hematol., 2005; 82: 100-107.
34. Levis M., et al.: A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood. 2002; 99: 3885-3891.
35. Weisberg E., et al.: Inhibition of mutant FLT3 receptors in leukemia cells by the smali molecule tyrosine kinase inhibitor PKC412. Cancer Cell., 2002; 1: 433-443.
36. Kelly L.M., et al.: CT53518, a novel selective FLT3 antagonist for the treatment of acute myelogenous leukemia (AML). Cancer CelI., 2002; 1: 421-432.
37. O´Farrell A.M., et al.: SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003; 101: 3597-3605.
38. Brown P., et al.: FLT3 inhibition selectively kills childhood acute lymphoblastic leukemia cells with high levels of FLT3 expression. Blood. 2005; 105: 812-820.
39. Piloto O., et al.: Inhibitory anti-FLT3 antibodies are capable of mediating antibody-dependent cell mediated cytotoxicity and reducing engraftment of acute myelogenous leukemia blasts in nonobese diabetic/severe combined immunodeficient mice. Cancer Res., 2005; 65: 1514-1522.
40. Piloto O., et al.: IMC-EB10, an anti-FLT3 monoclonal antibody, prolongs survival and reduces nonobese diabetic/severe combined immunodeficient engraftment of some acute lymphoblastic leukemia cell lines and primary leukemic samples. Cancer Res., 2006; 66: 4843-4851.
41. Smith B.O., et al.: Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004; 103: 3669-3676.
42. Stone R.M., et al.: Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005; 105: 54-60.
43. Smith B.O., et al.: Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004; 103: 3669-3676.
44. Stone R.M., et al.: Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005; 105: 54-60.
45. Fiedler W., et al.: A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005; 105: 986-993.
46. Levis M., et al.: In vitro studies of a FLT3 inhibitor combined with chemotherapy: sequence of administration is important to achieve synergistic cytotoxic effects. Blood. 2004; 104: 1145-1150.
47. Levis M., et al.: A randomized, open-label study of Lestaurtinib (CEP-701), an oral FLT3 inhibitor, administered in sequence with chemotherapy in patients with relapsed AML harboring FLT3 activating mutations: clinical response correlates with successful FLT3 inhibition. Blood. 2005; 106: 121a.
48. Stone R., et al.: Phase IB study of PKC412, an oral FLT3 kinase inhibitor, in sequential and simultaneous combinations with daunorubicin and cytarabine (DA) induction and high-dose cytarabine consolidation in newly diagnosed patients with AML. Blood. 2005; 1 06: 121 a.
49. Heidel F., et al.: Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006; 107:293-300.