© Borgis - Nowa Pediatria 4/2007, s. 97-100
Bartosz Godlewski1, Karol Jastrzębski2, Krzysztof Zapałowicz1
Fizjologiczne podstawy działania leków przeciwpadaczkowych
Physiological aspects of antiepileptic drugs mechanism of action
1Klinika Neurochirurgii i Chirurgii Nerwów Obwodowych USK nr 2 im. WAM w Łodzi
Kierownik: Prof. dr hab. n. med. Andrzej Radek
2Klinika Neurologii i Epileptologii USK nr 2 im. WAM w Łodzi
Kierownik: Prof. dr hab. n. med. Andrzej Klimek
Streszczenie
The main method of treatment of epilepsy is pharmacology. Pharmacology should be preceded be precise diagnostic. Mechanisms of action of antiepileptic drugs has been discovered relatively not long ago. Prior ignorance of these mechanisms of action didn´t limit their usefulness, but understanding the mechanisms helps in clinical practice. Thanks to this knowledge antiepileptic drugs can be used more rationally and effectively, especially in multidrug regimens. To understand and remember mechanisms of action it is necessary to have knowledge of physiology of nervous system. Many structures and processes are involved in the development of a seizure, including ion channels, neurotransmitters, receptors and synapses. The article includes information about physiology as well as pharmacology. The role of GABA (gamma-aminobutyric acid) receptors, sodium and potassium channels in generating of action potentials and in ethiopathology of seizures are described.

Większość żywych komórek utrzymuje stałą wartość różnicy potencjałów pomiędzy swym wnętrzem a otoczeniem. Ten stały potencjał wnętrza komórki względem jej otoczenia nazywamy potencjałem spoczynkowym. Zdolność komórki do utrzymywania stałej wartości potencjału spoczynkowego związana jest bezpośrednio z istnieniem różnicy stężeń niektórych jonów pomiędzy wnętrzem i otoczeniem komórki. Dla większości komórek jonami „najważniejszymi” z punktu widzenia potencjału spoczynkowego są jony sodu, potasu oraz chlorkowe. „Typowy” – czyli najczęściej spotykany – rozkład stężeń jonów jest taki, że na zewnątrz komórki stężenie jonów sodowych i chlorkowych jest większe niż wewnątrz komórki, natomiast stężenie jonów potasu jest większe wewnątrz komórki. Stała wartość potencjału błonowego może być utrzymywana jedynie wtedy, gdy całkowity ładunek przepływający przez błonę jest równy zeru (w przeciwnym razie następowałaby zmiana ładunku błony i związana z tym zmiana potencjału).
Niektóre z komórek, oprócz utrzymywania potencjału spoczynkowego są zdolne do szybkiej i krótkotrwałej zmiany potencjału błonowego – komórki pobudliwe. Komórki nie posiadające tej zdolności nazywamy komórkami niepobudliwymi. Chwilowa, impulsowa zmiana potencjału błony komórkowej nosi nazwę potencjału czynnościowego. Potencjał czynnościowy powstaje w komórce pobudliwej, gdy potencjał jej błony przekroczy pewną graniczną wartość nazywaną progiem pobudzenia. Warto zauważyć, że wielkość bodźca pobudzającego ma znaczenie jedynie dla powstania pojedynczego potencjału czynnościowego – nie ma ona natomiast wpływu na jego przebieg. Klasyczny potencjał czynnościowy składa się z kilku faz:
1) gwałtownego wzrostu potencjału błonowego (depolaryzacja),
2) nieco powolniejszego spadku potencjału błony (repolaryzacja),
3) okresu, gdy potencjał błony jest niższy od potencjału spoczynkowego (hiperpolaryzacja) (ryc. 1).
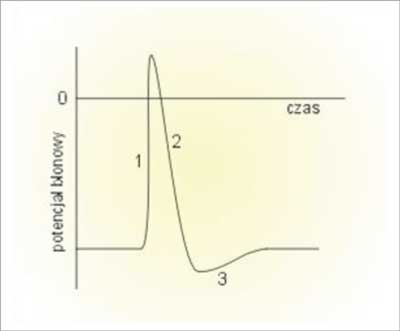
Ryc. 1. Zmiany potencjału błonowego podczas trwania potencjału czynnościowego w przykładowej komórce nerwowej: 1) depolaryzacja; 2) repolaryzacja; 3) hiperpolaryzacja.
W fazie depolaryzacji wzrost potencjału błony związany jest z napływem do wnętrza komórki dodatnich jonów. W czasie repolaryzacji ustaje dokomórkowy prąd sodowy (Na+), wzrasta natomiast odkomórkowy prąd potasowy (K+). Wypływ ładunków dodatnich powoduje zmniejszenie się potencjału błony komórkowej. Prąd potasowy płynie również wówczas, gdy potencjał błony osiąga wartość potencjału spoczynkowego – powoduje to, że komórka wchodzi w fazę hiperpolaryzacji. Dopiero, gdy ustanie prąd potasowy potencjał błony wraca do wartości spoczynkowej (1, 2).
Zdolność do kontrolowanego przepuszczania jonów zawdzięczamy kanałom jonowym. Ze względu na rodzaj czynnika aktywującego, kanały jonowe dzielimy na trzy zasadnicze grupy:
1) kanały zależne od napięcia,
2) kanały aktywowane chemicznie,
3) kanały aktywowane naprężeniem mechanicznym.
Bodziec pobudzający błonę o wartości ponadprogowej powoduje otwarcie odpowiednio dużej ilości kanałów sodowych i gwałtowny wzrost przewodnictwa błony dla jonów sodowych. Kanały sodowe po krótkim czasie ulegają inaktywacji i przewodnictwo błony dla sodu szybko maleje. Jednocześnie z kanałami sodowymi otwieraniu ulegają kanały potasowe – proces ten jest jednak wolniejszy i dlatego błona później osiąga maksymalną wartość przewodnictwa dla jonów potasu (ryc. 2.).
Ryc. 2. Przebieg zmian przepuszczalności błony dla jonów sodu i potasu w trakcie potencjału czynnościowego.
Mechanizm działania leków przeciwpadaczkowych na poziomie molekularnym polega na modulowaniu pobudliwości neuronów między innymi poprzez wpływ na kanały jonowe. Kanały sodowe i potasowe są miejscem uchwytu szeregu skutecznych leków przeciwpadaczkowych. Powstawanie potencjału czynnościowego związane jest również z neuroprzekaźnikami i ich receptorami. Neuroprzekaźniki służą do zmiany sygnału chemicznego na elektryczny, oddziaływują na kanały jonowe błony komórkowej aktywowane chemicznie. Na błonie postsynaptycznej występują receptory danego neuroprzekaźnika (receptory acetylocholiny, kwasu gamma-aminomasłowego (GABA), kwasu glutaminowego, dopaminy i wielu innych). Przyłączenie neuroprzekaźnika do błony postsynaptycznej powoduje zmianę jej polaryzacji (tzn. ujemnego potencjału elektrycznego wnętrza komórki postynaptycznej mierzonego względem przestrzeni zewnątrzkomórkowej). W przypadku neuroprzekaźnika o działaniu pobudzającym jest to zmiana dodatnia zwana depolaryzacją. W przypadku neuroprzekaźnika o działaniu hamującym jest to zmiana ujemna, zwana hiperpolaryzacją (3, 4, 5, 6).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Konturek S.: Fizjologia człowieka tom IV - neurofizjologia. Wyd. 6. Wydawnictwo Uniwersytetu Jagiellońskiego, Kraków 1998. 2. Traczyk W., Trzebski A.: Fizjologia człowieka z elementami fizjologii stosowanej i klinicznej. Wyd. 3. Wydawnictwo Lekarskie PZWL, Warszawa 2001. 3.Kurkowska-Jastrzębska I., i wsp.: Padaczka lekooporna a czynniki genetyczne. Farmakologia w Psychiatrii i Neurologii 2005; 1: 25-31. 4.Hirose S., et al.: Molecular genetics of human familial epilepsy syndromes. Epilepsia 2002; 43 (Suppl. 9): 21-25. 5.Matthews G.: Neurobiologia - od cząsteczek i komórek do układów. Wyd. 1. Wydawnictwo Lekarskie PZWL, Warszawa 2000. 6.Ganong W.: Fizjologia. Podstawy fizjologii lekarskiej. Wydawnictwo Lekarskie PZWL, Warszawa1994. 7.Jędrzejczak J., i wsp.: Choroby układu nerwowego. Wyd. 1. Wydawnictwo Lekarskie PZWL, Warszawa 2004, ss. 442-466. 8.Białecka M., i wsp.: Znaczenie polimorfizmu genu MDR-1 w patogenezie i leczeniu padaczki lekoopornej. Neurol. Neurochir. Pol., 2005; 39, 6: 476-481. 9.Glauser T.: Advancing the medical management of epilepsy: disease modification and pharmaetics. J. Child. Neurol., 2002; 17 (Suppl. 1): 85-93. 10.Smith P., Wallach S.: Leczenie padaczek. [W]: Padaczka - kliniczny przewodnik. Wyd. 1 polskie. ALFA - Medica Press, Bielsko-Biała 2003, ss. 128-164. 11.Goa K., et al.: Lamotrigine: a review of its pharmacological properties and clinica; efficacy in epilepsy. Drugs 1993; 2: 31-37. 12.Goldin A.: Mechanisms od sodium channel inactivation. Curr. Opin. Neurobiol., 2003; 3: 284-290. 13.Rhodes T., et al.: Nonivactivating voltage-gated sodium channels in severe myoclonic epilepsy of infancy. Proc. Natl. Acad. Sci., USA 2004; 101: 11147-11152. 14.Yamakawa K.: Epilepsy and sodium channel gene mitations: gain or loss of function? Neuroreport 2005; 16: 1-3. 15.Mc Lean M., et al.: Oxcarbazepine: mechanism of action. Epilepsia 1994; 35 (Suppl. 3): 5-9. 16.Jóźwiak S., i wsp.: Postępy w badaniach nad genetyką molekularną padaczek. Neurol. Neurochir. Pol., 2005; 39, 6: 497-508. 17.Meldrum B.: GABAergic mechanisms in the pathogenis and treatment of epilepsy. Br. J. Clin. Pharmacol., 1989; 27: 3-17. 18.Baulac S., et al.: First genetic evidence of GABA (A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat. Genet., 2001; 28: 46-48. 19.Ramakrishna L., Hess G. On the mechanism of a mutated and abnormally functioning gamma-aminoburytic acid (A) receptor linked to epilepsy. Biochemistry 2004; 43: 7534-7540. 20.Kulmann D., Hanna M.: Choroby neurologiczne spowodowane dziedzicznymi mutacjami kanałów jonowych. The Lancet Neurology PL 2003; 46: 629-637. 21.Chmielewska B., Stelmasiak Z.: Miejsce nowych leków przeciwpadaczkowych w farmakoterapii padaczki. Przew. Lek., 2001; 4, 11: 70-77. 22.Brodie M.: Tiagabine pharmacology in profile. Epilepsia 1995; 36 (Suppl. 6): 7-9. 23.Hill D., et al.: Autoradiographical detection of (3H) - gabapentin binding sites in rodent brain. Abstract. Br. J. Clin. Pharmacol., 1991; 104: 72. 24.Dichter M., Brodie M.: New antiepileptic drugs. N. Engl. J. Med., 1996; 334: 1583-1590.
