© Borgis - Postępy Nauk Medycznych 8/2008, s. 504-509
*Stanisław Zajączek
Siatkówczak
Retinoblastoma
Międzynarodowe Centrum Nowotworów Dziedzicznych Zakład Genetyki i Patomorfologii Pomorska Akademia Medyczna, Szczecin
Kierownik Zakładu: prof. dr hab. med. Jan Lubiński
Streszczenie
Siatkówczak jest modelowym nowotworem o podłożu genetycznym. Warunkowany jest dominującymi mutacjami jednej kopii genu RB1, które mogą mieć charakter somatyczny (S. sporadyczny, zwykle jednostronny) lub konstytucyjny (często obustronny i/lub wieloogniskowy). Do powstania guza konieczne jest jeszcze „drugie trafienie”, które ma już charakter mutacji somatycznej. Unieczynnienie obu kopii genu odblokowuje cykl komorkowy w fazie G1-S i inicjuje nowotwór. Gen RB1 składa się z 27 eksonów, mutacje nie wykazują preferencji lokalizacyjnych typu hot spot (co utrudnia diagnostykę), jednak ostatnio opisano pewne preferencje lokalizacyjne i etniczne w charakterystyce mutacji. Omówiono uwarunkowania genetyczne i charakterystykę rodowodowo-kliniczną siatkówczaka. Podano zasady diagnostyki genetycznej siatkówczaka a także zasady opieki nad rodzinami obciążonymi zmutowanym genem. Z uwagi na występowanie nosicieli mutacji konstytucyjnych również w grupie pacjentów z guzami jednostronnymi sporadycznymi, wszyscy chorzy z siatkówczakiem jak i ich rodzeństwo powinni być poddawani badaniom profilaktycznym do chwili potwierdzenia/wykluczenia mutacji. Nosicielstwo mutacji konstytucyjnej powoduje także w późniejszym życiu pacjenta podwyższone ryzyko innych pierwotnych nowotworów, zwłaszcza mięsaków.
Summary
Retinoblastoma genetics is modelling system for knowledge of basic features of all hereditary cancers. Tumour is caused by inactivating mutations of both copies of RB1 gene. They are transmitted as "one hit” in dominant manner, but inactivation of second gene copy is needed for initiation. In hereditary form (frequently bilateral/ multifocal) first "hit” is as constitutional and inherited but a second hit is a somatic mutations. In sporadic form (mostly unilateral and unifocal) both "hits” are somatic mutations. Inactivation of both RB1 gene copies abolish cell cycle block between G1 and S phase initialize carcinogenesis. RB1 gene consists of 27 eksons, mutations does not exhibit hot spots, but some spatial and ethnic regularities were detected in last years. Hereditary determinations, clinical value of pedigree and molecular analyses of RB1 gene are described, as well as basic rules of familial care. Due to detection of constitutional mutations carrier status in some patients with non-familial, unilateral tumours (de novo and germ- line or low penetrant mutations) prophylactic investigations of all first degree relatives must be performed, until their molecular verification is done. Patients with constitutional mutations have also higher risk of secondary primary tumours, particularly sarcomas.

Siatkówczak (S) zajmuje szczególne miejsce pośród nowotworów – jest to pierwszy nowotwór o udowodnionej dziedzicznej etiologii, na którego przykładzie kształtowały się idee „dwu trafień” i genów supresorowych (1, 2, 3).
S pojawia się z częstością 1: 25 000 żywych urodzeń. Mimo swojej rzadkości S jest najczęstszym nowotworem wewnątrzgałkowym u dzieci a większość przypadków rozpoznawanych jest przed 5 r.ż. U dorosłych S jest wielką rzadkością. 60% przypadków S ma charakter sporadyczny w ścisłym znaczeniu tego słowa; są one wywołane mutacjami somatycznymi w komórkach siatkówki. Pozostałe 40% pacjentów to dzieci obciążone mutacjami konstytucyjnymi – wśród nich 10-15% to przypadki rodzinne a pozostałych 25-30% to przypadki rodowodowo sporadyczne, powstałe jednak w wyniku konstytucyjnej mutacji germinalnej de novo. Mutacja konstytucyjna ma penetrację sięgającą 90%. Do powstania nowotworu konieczne jest unieczynnienie białka pRB; które ma miejsce dopiero po utracie funkcji obu kopii genu (4).
Prawdopodobieństwo powstania kolejnych dwu mutacji somatycznych genu RB1, następujących kolejno w tej samej komórce siatkówki – jak się to dzieje w S sporadycznym – jest wprost proporcjonalne do długości życia komórki; tłumaczy to fakt pojawiania się takich guzów w późniejszym wieku aniżeli S wywołanych mutacją konstytucyjną, zwykle także jednostronnie i jednoogniskowo (ryc. 1). Zgodnie z mechanizmem „dwu trafień” w guzach powstałych u osób z mutacją konstytucyjną, wobec jej istnienia już od urodzenia, do ostatecznego powstania nowotworu konieczna jest w komórce siatkówki już tylko pojedyncza mutacja somatyczna. Zdarzenie takie ma szansę zaistnieć wcześniej – stąd młodszy wiek chorych z S dziedzicznym. Druga mutacja – somatyczna, u nosiciela zmiany konstytucyjnej zdarzyć się może w większej liczbie komórek – stąd częsta wieloogniskowość i obustronność tych guzów (1, 3).
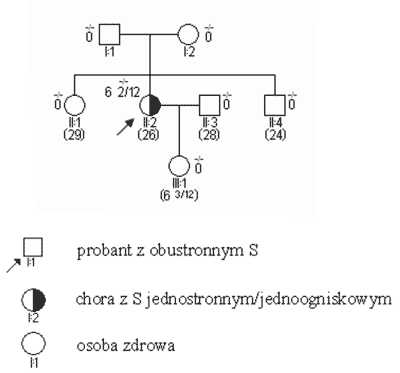
Ryc. 1. Rodowód w typowych uwarunkowaniach genetycznych, siatkówczak sporadyczny bez mutacji konstytucyjnej.
Legenda do rodowodów przedstawionych na rycinach 1-4:
–/– wykluczenie mutacji konstytucyjnej
+/– obecność mutacji konstytucyjnej jednego z alleli
3 2/12 wiek rozpoznania S w latach i miesiącach
(12) aktualny wiek w latach
S obustronne i jednostronne wieloogniskowe są zatem praktycznie zawsze związane z istnieniem mutacji konstytucyjnej, przekazanej rodzinnie lub nabytej de novo. Pojawiają się one w większości przypadków przed 3 r.ż. (ryc. 2). Mutacje konstytucyjne mogą jednak powstawać również jako zmiany germinalne d e novo (ryc. 3); związane z nimi guzy, pomimo iż rodowodowo sporadyczne, wykazują inne cechy kliniczne – w tym zwłaszcza wcześniejsze wystąpienie – podobne jak w nowotworach rodzinnych. Pewien odsetek chorych – jak wynika z naszych badań sięgający 20% – jest mimo jednostronności w chwili rozpoznania obciążony mutacją konstytucyjną (3, 5). Przypadki takie charakteryzują się (w stosunku do innych S jednostronnych) wcześniejszym wystąpieniem guza, wieloogniskowością i wysokim ryzykiem późniejszego pojawienia się nowego pierwotnego guza w tym samym lub drugim oku.
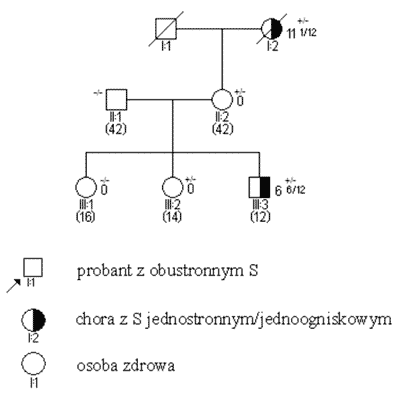
Ryc. 2. Rodowód w typowych uwarunkowaniach genetycznych, siatkówczak dziedziczny w wyniku mutacji konstytucyjnej o niepełnej penetracji.
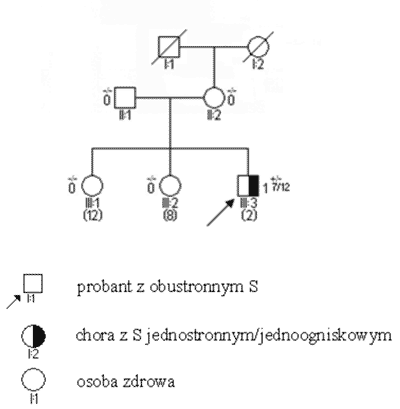
Ryc. 3. Rodowód w typowych uwarunkowaniach genetycznych, siatkówczak sporadyczny w wyniku mutacji konstytucyjnej de novo.
Gen i białko Retinoblastoma
Gen RB1 o genowo średnich rozmiarach (200 kB), zlokalizowany w prążku 13q14 składa się z promotora i 27 eksonów, znany jest jeden transkrypt RNA o rozmiarach 4,8 kB; zjawisko alternatywnego składania RNA nie dotyczy więc zapewne genu RB1.
Kodowane przez ten gen białko p105RB1 należy do grupy tzw. białek kieszeniowych i jest jądrową fosfoproteiną, wielkości 105-110 kDa. Uczestniczy ono w złożonych kaskadach regulacyjnych, decydujących o przejściu komórek w cyklu komórkowym z fazy G1 do fazy S a więc jest jednym z głównych regulatorów proliferacji. Dotyczy to wszystkich komórek ustroju a gen wykazuje znaczny konserwatyzm ewolucyjny. Białko oddziałuje na funkcję wielu innych genów przez unieczynnienie (sekwestrację) jednego z głównych regulatorów transkrypcji – czynnika E2F. Zdolność ta realizowana jest poprzez wiązanie E2F w „kieszeni” białka pRB1, uniemożliwiając czynnikowi E2F transaktywację innych podległych genów, co jest równoznaczne z utrzymaniem komórek w spoczynkowej fazie Go. Tylko białko pRB1 z hipofosforylowanymi resztami serynowymi posiada zdolność wiązania E2F; hiperfosforylacja białka uwalnia czynnik transkrypcyjny i umożliwia przejście G1 – S i progresję cyklu komórkowego. Do kieszeni białka pRB1 wiążą się kompetencyjnie również niektóre białka takich wirusów onkogennych jak białko E7 papillomawirusa, białko E1A adenowirusa, antygen T wirusa SV40 i inne. Zjawisko to pozwala powiązać w sposób niesprzeczny procesy ontogenezy wirusowej z istnieniem nowotworów genetycznie uwarunkowanych. Kieszeń białka pRB1 może wiązać również wiele innych białek zawierających motyw -Leu-x-Cys-x-Glu- (1, 4, 6, 7, 8). Rozpoznane u chorych z S mutacje sekwencji zlokalizowane są najczęściej w kieszeni białkowej i jej najbliższej okolicy i wpływają na powinowactwo do czynników transkrypcyjnych, ale niekiedy nie blokują całkowicie ich wiązania – mają wtedy cechy mutacji o niskiej penetracji (p. niżej).
Uniwersalne mechanizmy regulacyjne realizowane przez białko pRB1 mogą tłumaczyć udział jego mutacji w powstawaniu także innych niż S nowotworów, jak kostniakomięsak, rak pęcherza, rak drobnokomórkowy płuc i in., współwystępujących niekiedy rodzinnie z siatkówczakiem (9).
Aktywność genu RB1 jest regulowana na poziomie promotora przez przypisane mu regulatory transkrypcji; nieliczne poznane dotąd mutacje promotora upośledzają tą regulację i mają kliniczne cechy mutacji o niskiej penetracji (p. niżej). Jedną z nich – i pierwszą znaną de novo – opisaliśmy w naszym Ośrodku (10).
Dla prawidłowej funkcji komórek wystarczająca jest aktywność pojedynczej kopii genu; nosiciele mutacji konstytucyjnych poza predyspozycją do S nie różnią się w uchwytny sposób od posiadaczy dwu prawidłowych kopii genu.
Przebieg i charakterystyka kliniczna
Szczyt zachorowań na S występuje ok. 42 miesiąca życia, ponad 90% przypadków rozpoznawanych jest po raz pierwszy przed 5 r.ż. Znane są sytuacje, w których guz ten wykrywano tuż po urodzeniu. Wczesnymi objawami klinicznymi nasuwającymi podejrzenie S są: zez, przekrwienie i stan zapalny gałki ocznej. Zazwyczaj rozpoznanie guza następuje jednak na podstawie objawów później występujących; są nimi exophthalmos i zaobserwowane przez rodziców tzw. „kocie oko”- przeświecanie umiejscowionych na siatkówce serowatych mas guza przez soczewkę. Identyfikacja nowotworu w okresie wczesnym pozwala często na wyleczenie z ubytkami pola widzenia i zachowaniem gałki ocznej. Identyfikacja późniejsza, niestety najczęstsza, oparta o stwierdzenie „kociego oka” wiąże się zwykle z koniecznością usunięcia gałki ocznej oraz nierzadko uzupełniającej radio- i chemioterapii (3, 4).
Stosunkowo rzadko S daje klasyczne przerzuty a szerzy się głównie przez ciągłość drogą n. wzrokowego. Rokowanie zależy w znacznym stopniu od standardu diagnostyki i opieki medycznej; w krajach rozwiniętych o dobrej tradycji medycznej śmiertelność rzadko przekracza 8% a konieczność usunięcia gałki ocznej ma miejsce w ok. 10% przypadków. W krajach słabo rozwiniętych obserwowano śmiertelność 100%.
Podłoże molekularne dziedzicznego siatkówczaka
Predyspozycja dziedziczna do siatkówczaka wiąże się z istnieniem konstytucyjnych mutacji genu RB1.
Pełny zakres badań molekularnych tego genu dostępny jest w naszym Ośrodku (8, 10-14). Diagnostyka sekwencji genu obciążona jest znacznymi kosztami. Wykazano jednak, że koszty te, ze względu na wyłączanie nosicielstwa u wielu potencjalnie predysponowanych członków rodzin, są niższe aniżeli koszty pełnej opieki profilaktycznej koniecznej u wszystkich dzieci w rodzinie, jeśli nie dokonaliśmy wyłączeń (15). Pełne sekwencjonowanie genu RB1 identyfikuje mutację w ok. 80% rodzin, w których S ma ewidentnie dziedziczny charakter. Pewnym ułatwieniem może być wstępne stosowanie metod preselekcyjnych jak SSC, DGGE, PTT; mają one jednak niższą czułość aniżeli sekwencjonowanie. Identyfikacja mutacji u osoby chorej pozwala następnie na weryfikację tylko wybranego fragmentu genu o pozostałych członków rodziny (4, 16). W diagnostyce wykorzystać można także analizy sprzężeń z wykorzystaniem molekularnych markerów wewnątrzgenowych; są one pomocne zwłaszcza w diagnostyce większych liczebnie rodzin (4, 10, 13, 17). Do diagnostyki S wdrożono nie tylko klasyczne PCR – sekwencjonowanie „ekson po eksonie” (17-20), ale także techniki takie jak multiplex i jakościowy multiplex -PCR (umożliwiające jednoczesną ocenę kilku eksonów „pakiecie”), jak i nowe techniki końcowej detekcji mutacji wywodzące się z biochemii np. HPLC (13, 14, 19, 21).
Źródłem diagnostyki DNA konstytucyjnego jest zwykle krew obwodowa a analizie podlega pozyskany z niej DNA. Materiał ten zawiera zarówno kodujące (eksony) jak i niekodujące (introny) części genu. Stosowanie jako materiału badanego RNA i przygotowanego in vitro na jego matrycy cDNA, zawierającego jedynie kodujące fragmenty DNA, ułatwia zarówno technikę badania jak i jego późniejszą interpretację (16). Diagnostyka DNA samego guza nie jest konieczna do identyfikacji mutacji konstytucyjnych, może jednak być potwierdzeniem prawidłowości procedur i źródłem cennych informacji o mutacjach somatycznych („drugich trafieniach” i „sporadycznych”).
Informacje o zaobserwowanych mutacjach gromadzone są w dwu bazach danych z wolnym dostępem on-line (18, 21). Baza danych Dr Lohmanna obejmuje mniejszą liczbę mutacji i jest obecnie rzadziej aktualizowana, umożliwia natomiast łatwe różnicowanie mutacji konstytucyjnych od somatycznych „drugich trafień”; w bazie Valverde i wsp., zawierającej więcej nowszych danych (932 mutacje), nie zawsze takie rozróżnienie jest możliwe. Jednakże właśnie analizy materiału tej drugiej bazy danych radykalnie zmieniły naszą wiedzę o charakterystyce konstytucyjnych mutacji genu RB1.
Uważano do niedawna, że mutacje te zlokalizowane są bez wyraźnych prawidłowości (16). Jak się jednak okazało w genie RB1 występują „gorące miejsca” nagromadzenia mutacji, odpowiadające czynnościowo ważnym okolicom białka pRB1 a zwłaszcza jego „kieszeni” – 79% powtarzających się mutacji to tranzycje C/T w 11 trypletach kodonów argininowych CGA z czego aż 40% jest zawartych w eksonach 8, 10, 11, 14, 15, 17, 18 i 23 (22). Obserwacje te znacząco modyfikują taktykę sekwencjonowania diagnostycznego, jego szybkość i koszty (21).
Przeważająca większość mutacji to, jak się spodziewano, zmiany uniemożliwiające skuteczną syntezę białka, bądź to unieczynniające je funkcjonalnie; są to zwłaszcza mutacje typu „stop” (42% obserwowanych), ale także dodatkowo niektóre mutacje typu delecji i „splicing site”.
W odróżnieniu od mutacji „inaktywujących”, mutacje modyfikujące funkcjonalność cząsteczki, ale nieblokujące całkowicie syntezy białka pRB są rozmieszczone losowo.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. DiCiomino D, Gallie B, Bremner R: Retinoblastoma: The disease, Gene and Protein Provide Critical Lead to Understand Cancer. Cancer Biol 2000, 10: 255-269.
2. Gallie BL, Ellsworth RM, Abramson DH, Philips RA: Retinoma: Spontaneous Regression of Retinoblastoma or Benign Manifestation of Mutation? Br J Cancer 1982, 45: 513-521.
3. Murphee AL, Clark RD: Retinoblastoma. In: Emery´s and Rimoin´s Principles and Practice of Medical Genetics. D.L.Rimoin, J.M.Connor, R.E.Pyeritz, B.R. Korf (eds), Churchill – Livingstone, London 2002, 3: 3604-3636.
4. Gallie B, et al.: The Genetics of the Retinoblastoma: Relevance to the Patient. Paed Clin North Am 1991, 38: 299-315.
5. Munier FL, et al.: Evidence of Somatic and Germinal Mosaicism in Pseudo-Low Penetrant Hereditary Retinoblastoma, by Constitutional and Single Sperm Mutation Analysis. Am J Hum Genet 1998, 63: 1903-1908.
6. Gutkind JS (Ed.) Signalling Networks and Cell Cycle Control: The Molecular Basis of Cancer and other Diseases. Humana Press, Champaing, IL, 2000.
7. Xiao B, et al.: Crystal Structure of the Retinoblastoma Tumor Supressor Protein Bound to E2f and the Molecular Basis of its Regulation. Proc Natl Acad Sci USA 2003, 100.
8. Zajączek S: Ocena konstytucyjnych mutacji genu Rb-1 i niestabilności chromosomów w diagnostyce dziedzicznych postaci i badaniach patogenezy siatkówczaka jednostronnego. Praca habilitacyjna. Pomorska Akademia Medyczna w Szczecinie 1999, 1-80.
9. Abramson DH: Second Non-Ocular Cancers in Retinoblastoma: A Unified Hypothesis, Ophth Genet 1999, 20: 193- 203.
10. Zajączek S, et al.: Frequency and Nature of germline RB-1 Gene Mutations In a Series of Patients with Sporadic Unilateral Retinoblastoma. Eur J Cancer 1999, 35: 1824-1827.
11. Jakubowska A, et al.: Novel RB1 Gene Constitutional Mutations Fund In Polish patients with familial and/or Bilateral Retinoblatoma, Hum Mutat 2001, 18: 459 – Online Mutations in Brief #456.
12. Zajączek S, et al.: Age At Diagnosis to Discriminate those Patients for whom Constitutional DNA Sequencing is appropriate in Sporadic Unilateral Retinoblastoma. Eur J Cancer 1998, 34: 1919-1921.
13. Zajączek S: Genetics of Retinoblastoma in Clinical Practice. Ann Diagn Paed Pathol 2003, 3: 35-39.
14. Zajączek S: Genetyka siatkówczaka w praktyce klinicznej – przegląd nowych zagadnień. Okulistyka 2007, 10: 7-13.
15. Noorani HZ, Khan HN, Gallie BL, Detsky AS: Cost Comparison of Molecular vs Conventional Screening of Relatives at Risk for Retinoblastoma. Am J Hum Genet 1996, 59: 301-307.
16. Harbour WJ: Overview of RB Gene Mutations in Patients with Retinoblastoma. Implications for Clinical Genetics. Ophthalmology 1998, 105: 1442-1447.
17. Lohmann DR, et al.: The Spectrum of RB1 Germline Mutations in Hereditary Retinoblastoma, Am J Hum Genet 1996, 58: 940-949.
18. Lohmann DR, et al.: Retinoblastoma Gene Clinics 2003: http://www.geneclinics.org/profiles/retinoblastoma/details.html
19. Richter S, et al.: Sensitive and Efficient Detection of RB1 Gene Mutations Enhances Care for Families with Retinoblastoma. Am J Hum Genet 2003, 72: 253-269.
20. Otterson G, et al.: Temperature Sensitive RB Mutations Linked to Incomplete Penetrance of Familial Retinoblastoma in 12 Families. Am J Hum Genet 1999, 65: 1040-1046.
21. Valverde JR, Alonso J, Palacios I, Pestana A: RB1 Gene Mutation Up-to Date: a Meta-Analysis Based on 932 Reported Mutations Available in a Searchable Database. BMC Genetics 2005, 6: 1-9, http://www.biomedcentral.com/1471-2156/6/53.
22. Lohmann DR, et al.: Constitutional RB1 Gene Mutations in Patients with Isolated Unilateral Retinoblastoma. Am J Hum Genet 1997, 61: 282-294.
23. Schubert EL, Strong LC, Hansen MF: A Splicing Mutation in RB1 in Low Penetrance Retinoblastoma. Hum Genet 1997, 100: 557-563.
24. Scheffer H, et al.: Linkage Analysis of Families with Hereditary Retinoblastoma: Nonpenetrance of Mutation. Am J Hum Genet 1989, 45: 252-260.
25. Klutz M, Bockmann D, Lohmann DR: A Parent-of -Origin Effect in Two Families with Retinoblastoma is Associated with a Distinct Splice Mutation in the RB1 Gene. Am J Hum Genet 2002, 71: 174-179.
26. Munier FL, et al.: Evidence of Somatic and Germinal Mosaicism in Pseudo-Low Penetrant Hereditary Retinoblastoma, by Constitutional and Single Sperm Mutation Analysis. Am J Hum Genet 1998, 63: 1903-1908.
27. Cowell JK, Gallie BL: Which Retinoblastoma Patients Should be Screened for RB1 Mutations? Eur J Cancer 1998, 34: 1825-1826.
28. National Cancer Institute, 2006 – Retinoblastoma Treatment: http://www.cancer.gov/cancertopics/pdq/treatment/ retinoblastoma/healthprofessional
