Jacek Skiendzielewski1, Beata Kucińska1, Danuta Roik2, *Bożena Werner1
Problemy diagnostyczno-terapeutyczne u noworodka z zespołem Turnera – opis przypadku
The newborn with Turner syndrome, diagnostic and therapeutic considerations – a case report
1Klinika Kardiologii Wieku Dziecięcego i Pediatrii Ogólnej, Warszawski Uniwersytet Medyczny
Kierownik Kliniki: prof. dr hab. n. med. Bożena Werner
2Zakład Radiologii Pediatrycznej, Warszawski Uniwersytet Medyczny
Kierownik Zakładu: dr n. med. Michał Brzewski
Summary
Turner syndrome is a chromosomal disorder caused by complete or partial X chromosome monosomy that manifests various clinical features depending on the karyotype and on the genetic background of affected girls. The most prevalent karyotype is 45,X, followed by mosaic patterns. Turner syndrome is associated with several morbidities that increase with age. Turner syndrome causes typical dysmorphic features (webbed neck, lymphedema of the hands and feet, narrow and high-arched palate, low hairline at the back, low set ears, broad chest and wide set nipples) and several multisystem disorders: short stature, primary gonadal deficiency, kidney malformations. It has been reported that the 50% of adult patients and 30% of pediatric patients with Turner syndrome present with cardiovascular abnormalities. The authors present a case of a newborn with Turner syndrome, coarctation of aorta and bicuspid aortic valve. The child was operated on coarctation of aorta shortly after the diagnosis. The regular check-ups showed good surgical result.

Wstęp
Zespół Turnera (ang. Turner syndrome – TS) występuje z częstością ok. 1:2500 noworodków płci żeńskiej. Najczęściej jest wynikiem monosomii chromosomu X. Dziewczynki dotknięte zespołem mają najczęściej kariotyp 45,X (ponad 50% przypadków), stosunkowo często spotyka się także izochromosom długich ramion chromosomu X, delecję krótkiego ramienia jednego z chromosomów X, mozaikowatość. Zespół charakteryzują typowe cechy dysmorfii: trójkątny kształt twarzy, niska linia owłosienia na czole i karku, skośne ustawienie szpar powiekowych, gotyckie podniebienie, nisko osadzone uszy, niedorozwój żuchwy, płetwiasta szyja, puklerzowata klatka piersiowa z szeroko rozstawionymi brodawkami, koślawość łokci, niedorozwój paznokci. Pacjentki z TS wymagają wielospecjalistycznej opieki, gdyż poza charakterystycznym fenotypem obserwuje się: niski wzrost, pierwotny brak miesiączki, bezpłodność, ponadto choroby układu krążenia (50%), choroby nerek (ok. 30-40%), nadciśnienie tętnicze (20%), choroby autoimmunologiczne (chorobę Hashimoto, celiakię, cukrzycę typu I), zaburzenia słuchu, wady wzroku. Choroby układu sercowo-naczyniowego mają największy wpływ na rokowanie w tej grupie pacjentów.
Opis przypadku
Trzytygodniowy noworodek został skierowany do oddziału kardiologii z przyszpitalnej poradni kardiologicznej z powodu podejrzenia zwężenia cieśni aorty (ang. coarctation of the aorta – CoA). Dziecko z ciąży 1, porodu 1, wiek matki 24 lata, w rutynowo wykonanym USG w ciąży stwierdzono wodniak karku, poza tym bez nieprawidłowości. W 25. tygodniu ciąży wykonano kordocentezę, stwierdzono nieprawidłowy kariotyp płodu 45,X. Noworodek urodzony w 37. tygodniu, z masą ciała 3560 g, długość 50 cm, oceniony na 7-7-8-8 punktów w skali Apgar. W badaniu przedmiotowym stwierdzano rozległe obrzęki stóp i podudzi, płetwiastą szyję, szmer skurczowy u podstawy serca. Noworodek wymagał początkowo wsparcia oddechowego poprzez nCPAP, od 2. doby oddech własny z tlenoterapią bierną do 5. doby. Z uwagi na zaburzenia oddychania, nie mogąc wykluczyć zakażenia uogólnionego, stosowano empiryczną antybiotykoterapię (ampicylina z gentamycyną) do czasu uzyskania ujemnych wyników wykładników stanu zapalnego oraz posiewu krwi. W 4. dobie wykonano przezklatkowe badanie echokardiograficzne: stwierdzono zwężenie w miejscu cieśni aorty do ok. 3 mm, niespełniające kryteriów rozpoznania CoA (stosunek wymiaru cieśni aorty do średnicy aorty brzusznej na poziomie przepony był > 50%), prawidłowy przepływ w aorcie brzusznej, prawidłową wielkość jam serca i kurczliwość lewej komory serca; ponadto dwupłatkową zastawkę aortalną (ang. bicuspid aortic valve – BAV) bez zaburzeń jej funkcji (zwężenia czy niedomykalności), przetrwałą lewą żyłę główną górną (wariant anatomiczny), niewielki lewo-prawy przeciek krwi w miejscu otworu owalnego i przewodu tętniczego (norma dla wieku).
W trakcie obserwacji w warunkach oddziału neonatologicznego stan noworodka pozostawał stabilny, przybierał na wadze, początkowo stosowano częściowe żywienie pozajelitowe, następnie karmiony butelką mlekiem początkowym. Wykonano szczepienia, przesiewowe testy metaboliczne i przesiewowe badanie słuchu (wynik prawidłowy). Ciśnienie tętnicze oraz saturacja na prawej kończynie górnej i prawej kończynie dolnej były prawidłowe. W kontrolnym echo serca po tygodniu bez progresji zaburzeń hemodynamicznych. Wypisany z oddziału noworodkowego w 14. dobie życia z zaleceniami kontroli po tygodniu w poradni kardiologicznej oraz planowej wizyty w poradni genetycznej, endokrynologicznej i audiologicznej.
Po tygodniu, zgodnie z zaleceniami rodzice zgłosili się z dziewczynką do poradni kardiologicznej. Niepokój kardiologa wzbudziło bardzo słabo wyczuwalne tętno na tętnicach udowych. Z podejrzeniem CoA skierował dziecko do szpitala w trybie pilnym.
Przy przyjęciu do oddziału kardiologii noworodek w stanie ogólnym dobrym, z fenotypowymi cechami TS (typowa dysmorfia twarzy, obrzęki limfatyczne stóp, płetwiasta szyja, niska linia owłosienia na karku). W badaniu układu krążenia bez cech niewydolności serca, szmer skurczowy 2/6 w skali Levine’a u podstawy serca promieniujący do okolicy międzyłopatkowej, bardzo słabo wyczuwalne tętno na tętnicach kończyn dolnych, ciśnienie tętnicze na prawej kończynie górnej 96/47 mmHg, na prawej kończynie dolnej 65/35 mmHg.
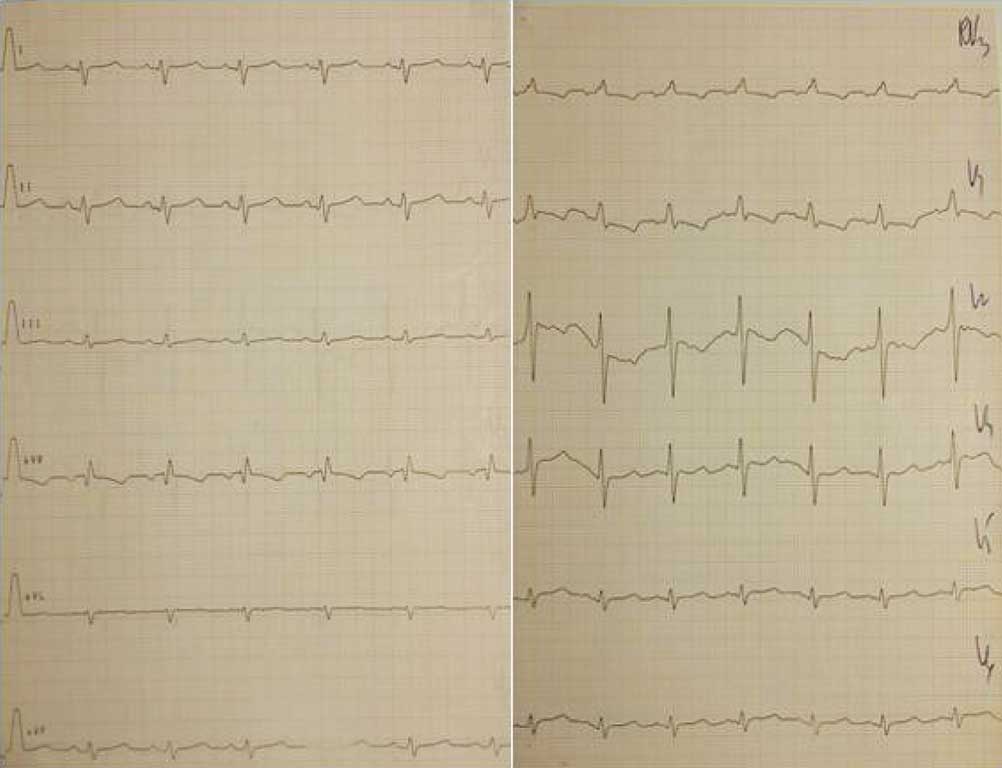
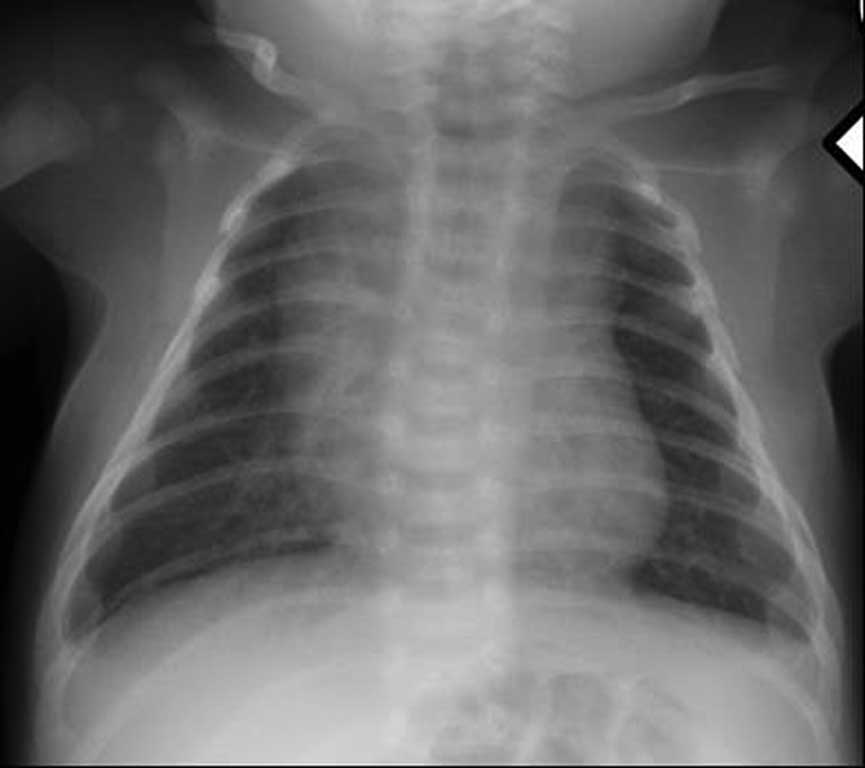
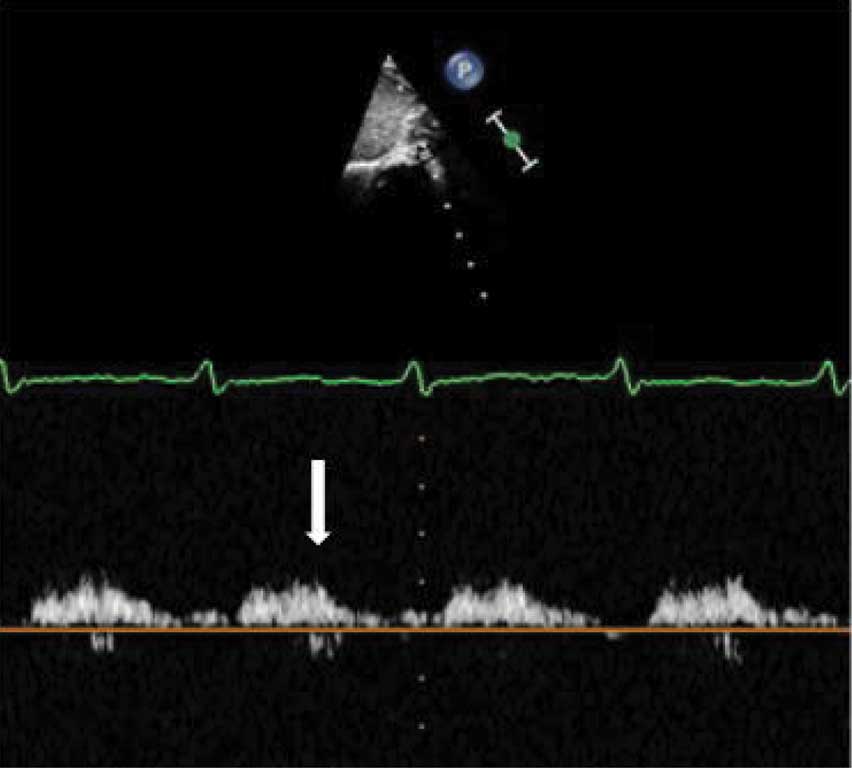
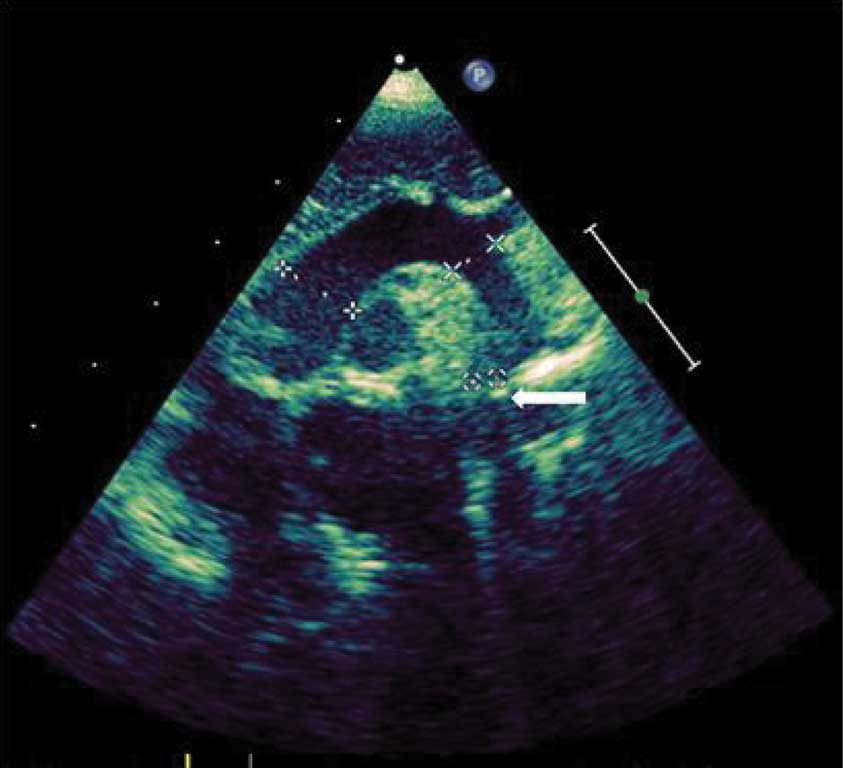
W badaniach laboratoryjnych krwi (morfologia krwi, jonogram, biochemia, hormony tarczycy) nie stwierdzono istotnych nieprawidłowości. Zapis 12-odprowadzeniowego ekg (ryc. 1) i zdjęcia przeglądowe klatki piersiowej (ryc. 2) były prawidłowe. USG jamy brzusznej uwidoczniło zdwojenie układu kielichowo-miedniczkowego nerki lewej, USG przez-ciemiączkowe było w normie, z kolei USG szyi uwidoczniło po stronie lewej przestrzeń płynową o wymiarach 33 x 15 x 29 mm, odpowiadającą obrazowi naczyniaka limfatycznego. W echo serca przepływ w aorcie brzusznej z zachowaną pulsacją, ale wydłużony na fazę rozkurczu (ryc. 3), aorta brzuszna na poziomie przepony o średnicy 6 mm. Cieśń w najwęższym miejscu do ok. 2 mm (ryc. 4), z nieprawidłowym kształtem przepływu krwi (ryc. 5), z maksymalnym gradientem ciśnienia skurczowego przez miejsce zwężenia do 60 mmHg. Zastawka aortalna dwupłatkowa, o prawidłowej funkcji. Pierścień aortalny ok. 8 mm, opuszka aorty 10 mm, aorta wstępująca 9,4 mm, część poprzeczna łuku 6 mm, aorta zstępująca za zwężeniem ok. 7 mm. Wielkość i funkcja skurczowa lewej komory serca były w normie. Rejestrowano śladowy lewo-prawy przepływ krwi przez otwór owalny, nie zarejestrowano przepływu przez przewód tętniczy, obserwowano wyraźnie widoczny przepływ w naczyniach międzyżebrowych odchodzących od aorty przed zwężeniem.

Ryc. 1. Dwunastoodprowadzeniowy zapis ekg, przesuw 50 mm/s, cecha 10 mm/mV. Zapis w normie dla wieku – prawogram, dość niski woltaż zespołów QRS w odprowadzeniach kończynowych

Ryc. 2. Przeglądowe zdjęcie klatki piersiowej. Wynik prawidłowy. Grasica tworzy cień śródpiersia górnego – w normie dla wieku
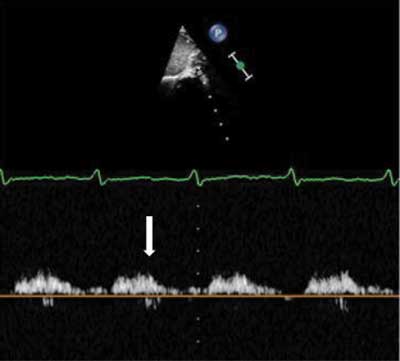
Ryc. 3. Nieprawidłowy kształt krzywej przepływu w aorcie brzusznej w badaniu dopplerowskim metodą fali pulsacyjnej – fala przepływu spłaszczona i wydłużona na fazę rozkurczu (strzałka)
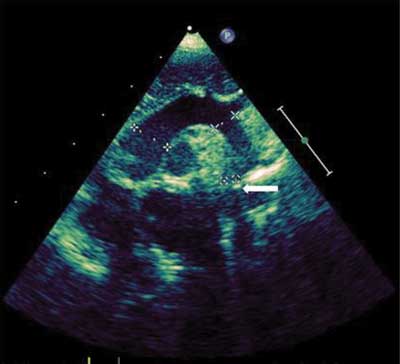
Ryc. 4. Wizualizacja łuku aorty echokardiografią dwuwymiarową w projekcji nadmostkowej – pomiary kolejno od strony lewej aorty wstępującej, części poprzecznej łuku aorty oraz cieśni aorty (strzałka)

Ryc. 5. Tzw. „zęby piły” – charakterystyczny profil przepływu krwi w miejscu zwężenia cieśni aorty oceniany w badaniu dopplerowskim metodą fali ciągłej
Celem pełnej wizualizacji anomalii naczyniowej wykonano angiotomografię komputerową łuku aorty we śnie spontanicznym, potwierdzając ostatecznie rozpoznanie CoA (ryc. 6).
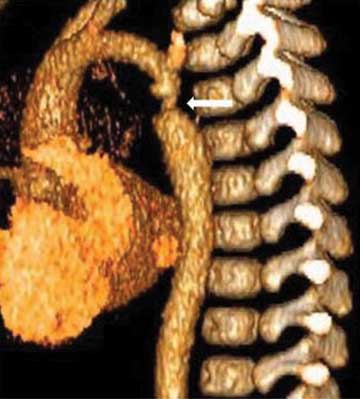
Ryc. 6. Ocena anatomii łuku aorty metodą angiotomografii komputerowej. Strzałka wskazuje zwężoną cieśń aorty
Na podstawie obrazu klinicznego, echokardiograficznego i tomografii komputerowej rozpoznano CoA. Ponadto w badaniu echo stwierdzono BAV bez zaburzeń funkcji zastawki aortalnej, bez poszerzenia opuszki aorty ani aorty wstępującej. Na konsylium kardiochirurgicznym dziewczynkę zakwalifikowano do operacji CoA. Przeniesiono ją do oddziału kardiochirurgicznego, gdzie w 30. dobie życia wykonano operację. W pooperacyjnym badaniu echokardiograficznym: przepływ o prawidłowym spektrum w miejscu operowanym, prawidłowa wielkość i kurczliwość lewej komory serca, prawidłowy przepływ w aorcie brzusznej. W trakcie obserwacji w oddziale kardiochirurgii bez niepokojących objawów, prawidłowe tętno na tętnicach kończyn dolnych, ciśnienie tętnicze na prawej kończynie górnej 97/50 mmHg, na prawej kończynie dolnej 95/54 mmHg. Wypisana do domu w 11. dobie po operacji w stanie dobrym, z zaleceniem dalszej opieki wielospecjalistycznej, bez leków.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Turner HH: A syndrome of infantilism, congenital webbed neck, and cubitus valgus. Endocrinology 1938; 28: 566-574.
2. Alpman A, Cogulu O, Akgul M et al.: Prenatally diagnosed Turner syndrome and cystic hygroma: incidence and reasons for referrals. Fetal Diagn Ther 2009; 25: 58-61.
3. Bondy CA, Turner Syndrome Study Group: Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group. J Clin Endocrinol Metab 2007; 92: 10-25.
4. Scholl J, Chasen ST: First trimester cystic hygroma: does early detection matter? Prenat Diagn 2016; 36: 432-436.
5. Hagman A, Wennerholm UB, Kallen K et al.: Women who gave birth to girls with Turner syndrome: maternal and neonatal characteristics. Hum Reprod 2010; 25: 1553-1560.
6. Yeşilkaya E, Bereket A, Darendeliler F et al.: Turner syndrome and associated problems in Turkish children: a multicenter study. J Clin Res Pediatr Endocrinol 2015; 7: 27-36.
7. Mortensen KH, Andersen NH, Gravholt CH: Cardiovascular phenotype in Turner syndrome – integrating cardiology, genetics, and endocrinology. Endocr Rev 2012; 33: 677-714.
8. Floriańczyk F: Wady łuku aorty. [W:] Werner B (red.): Wady serca u dzieci dla pediatrów i lekarzy rodzinnych. Wyd. I. Medical Tribune Polska, Warszawa 2015: 155-173.
9. Han BK, Rigsby CK, Hlavacek A et al.: Computed Tomography Imaging in Patients with Congenital Heart Disease Part I: Rationale and Utility. An Expert Consensus Document of the Society of Cardiovascular Computed Tomography (SCCT): Endorsed by the Society of Pediatric Radiology (SPR) and the North American Society of Cardiac Imaging (NASCI). J Cardiovasc Comput Tomogr 2015; 9: 475-492.
10. Korbmacher B, Krogmann ON, Rammos S et al.: Repair of critical aortic coarctation in neonatal age. J Cardiovasc Surg 2002; 43: 1-6.
11. King H, Waldhausen JA: Aortoplasty in infants with coarctation. Circ 1963; 27: 890-894.
12. Thanopoulos BD, Giannakoulas G, Giannopoulos A: Initial and six-year results of stent implantation for aortic coarctation in children. Am J Cardiol 2012; 109: 1499-1503.
13. Lucas V: Stent treatment of neonatal coarctation: another option for critically ill or extremely small patients with unoperated coarctation or failed surgery. Catheter Cardiovasc Interv 2010; 75: 562.
14. Ammar RI: Balloon angioplasty for native aortic coarctation in children and infants younger than 12 months: immediate and medium-term follow-up. J Invasive Cardiol 2012; 12: 662-666.
15. Agnoletti G, Bonhoeffer P, Borghi A et al.: Age-related aspects of balloon angioplasty for postsurgical aortic recoarctation. Cardiol Young 2002; 12: 470-473.
16. Prowotorow-Iwaniukowicz A, Skiendzielewski J, Pietrzak R, Werner B: Analiza wybranych parametrów klinicznych i echokardiograficznych u dzieci z dwupłatkową zastawką aortalną. Nowa Ped 2015; 19: 50-56.
17. Carro A, Teixido-Tura G, Evangelista A: Aortic dilatation in bicuspid aortic valve disease. Rev Esp Cardiol 2012; 65: 977-981.
18. Bilge I, Kayserili H, Emre S et al.: Frequency of renal malformations in Turner syndrome: analysis of 82 Turkish children. Pediatr Nephrol 2000; 14: 1111-1114.
19. Fudge EB, Constantacos C, Fudge JC, Davenport M: Improving detection of hypertension in girls with turner syndrome using ambulatory blood pressure monitoring. Horm Res Paediatr 2014; 81: 25-31.
20. Matura LA, Ho VB, Rosing DR, Bondy CA: Aortic dilatation and dissection in Turner syndrome. Circulation 2007; 116: 1663-1670.
21. Carlson M, Airhart N, Lopez L, Silberbach M: Moderate aortic enlargement and bicuspid aortic valve are associated with aortic dissection in Turner syndrome: report of the international turner syndrome aortic dissection registry. Circulation 2012; 126: 2220-2226.
22. Baena N, De Vigan C, Cariati E et al.: Turner syndrome: evaluation of prenatal diagnosis in 19 European registries. Am J Med Genet A 2004; 129A: 16-20.
23. Dalla Pozza R, Bechtold S, Urschel S et al.: QTc interval prolongation in children with Turner syndrome: the results of exercise testing and 24-h ECG. Eur J Pediatr 2009; 168: 59.
24. Kim HK, Gottliebson W, Hor K et al.: Cardiovascular anomalies in Turner syndrome: spectrum, prevalence, and cardiac MRI findings in a pediatric and young adult population. Am J Roentgenol 2011; 196: 454-460.