Joanna Chorostowska-Wynimko, Ewa Sommer, Ewa Rogala
Nowe perspektywy zastosowania niesteroidowych leków przeciwzapalnych w terapii chorób nowotworowych
New perspectives of non-steroidal anti-inflammatory drugs appliance in cancer treatment
z Zakładu Immunologii, Instytutu Gruźlicy i Chorób Płuc w Warszawie
Kierownik Zakładu: prof. dr hab. med. Ewa Skopińska-Różewska
Streszczenie
The paper contains a general review of current opinios on the perspectives of antiinflammatory non-steroid drugs (NSAIDs) appliance in the cancer treatment. The results of experimental, clinical and epidemiological studies are discussed. Author presents as well hypothetical mechanisms responsible for NSAIDs antineoplastic properties.

Niesteroidowe leki przeciwzapalne (NLPZ) są jedną z najczęściej przepisywanych przez lekarzy i bardzo chętnie stosowanych przez pacjentów grup leków. Powszechnie znane są ich właściwości przeciwbólowe, przeciwgorączkowe oraz przeciwzapalne. Najnowsze badania naukowe wskazują jednak, że wymienione korzystne działania NLPZ nie wyczerpują ich potencjału terapeutycznego. Coraz więcej danych doświadczalnych i epidemiologicznych potwierdza bowiem, że NLPZ wykazują również znaczące z klinicznego punktu widzenia właściwości przeciwnowotworowe.
Pierwsze przesłanki świadczące o możliwych niestandardowych zastosowaniach NLPZ pojawiły się już przed dwudziestu laty w pracach doświadczalnych prowadzonych na zwierzętach. Pollard i Lucket w 1983 roku wykazali, że przewlekłe podawanie indometacyny ma u szczurów laboratoryjnych działanie ochronne w modelu doświadczalnie (chemicznie) indukowanego raka jelita grubego. U zwierząt, którym podawano lek przez kilkanaście tygodni stwierdzano znacząco niższą liczbę i mniejsze rozmiary guzów nowotworowych (1). Zbliżone wyniki uzyskali autorzy badający w podobnych układach inne NLPZ: piroxicam (2), sulindak (3), ibuprofen oraz aspirynę (4). Co więcej z badań Rao i wsp. wynika, że supresyjne działanie NLPZ wykazują zarówno wobec postaci inwazyjnej, jak i nieinwazyjnej nowotworu (3). Znaczne ograniczenie tempa wzrostu guza pod wpływem indometacyny, aspiryny i meloxicamu uzyskano również u zwierząt badanych w odmiennych schematach doświadczalnych, gdy myszom wszczepiano komórki nowotworowe włókniakomięsaka, czy też gruczolakoraka jelita grubego (5, 6).
W zaprezentowanych pracach hamujący wpływ NLPZ na rozwój tkanki nowotworowej był zależny od zastosowanej dawki leku oraz momentu jego zadziałania. W pracy Moorghen´a i wsp. sulindak hamował u myszy powstawanie chemicznie indukowanego raka okrężnicy jedynie, gdy był podawany przez cały okres trwania doświadczenia, a więc od momentu zastosowania karcinogenu. Włączony później, po 17 tygodniach od rozpoczęcia chemicznej indukcji nowotworu, nie powodował zmniejszenia wzrostu i spowolnienia rozwoju guza (7). W przypadku piroxicamu nie obserwowano tak silnej zależności od czasu podania leku. Lek ten wykazywał działanie ochronne podany nawet w 13 tygodni po zastosowaniu karcinogenu, zmniejszając częstość występowania i ilość guzów jelita w stopniu zależnym od dawki (2). Wydaje się, że przytoczone wyniki stanowią przesłankę świadczącą o hamowaniu przez NLPZ już bardzo wczesnych etapów formowania się tkanki nowotworowej.
Wkrótce po opublikowaniu wyników pierwszych prac doświadczalnych na zwierzętach rozpoczęto badania kliniczne, których celem była ocena faktycznej przydatności terapeutycznej tych niestandardowych właściwości NLPZ. Już w 1983. Waddell i Loughry zastosowali sulindak u czterech pacjentów z rodzinną wielopolipowatością jelita grubego (familiar adenomatous poliposis), uzyskując po rocznym leczeniu całkowitą regresję zmian w jelitach (8). Podobne efekty obserwowali po zakończeniu pięcioletniego okresu leczenia (9). Wykazali również, że przerwanie terapii wiązało się z nawrotem choroby, potwierdzając w ten sposób kluczową rolę sulindaku w uzyskaniu korzystnego efektu klinicznego.
W późniejszych badaniach prowadzonych z zastosowaniem placebo i podwójnie ślepej próby Labayle i wsp. zastosowali sulindak (300 mg/dz) przez okres 6-12 miesięcy u 9 pacjentów z mnogą polipowatością, uzyskując w 6 przypadkach całkowitą, a w trzech częściową regresję zmian (10). Podobnie Giardello i wsp. u 22 pacjentów z tą jednostką chorobową przyjmujących sulindak przez 9 miesięcy obserwowali redukcję liczby polipów u 44% chorych, a zmniejszenie ich rozmiarów u 35% (11). Powyższe badania potwierdziły również znaczenie przed- i pooperacyjnej terapii sulindakiem u chorych z zaawansowanym stadium polipowatości jelita grubego.
Należy dodać, że kliniczne efekty stosowania sulindaku w przypadkach pojedynczych polipów jelita nie są tak jednoznaczne (12, 38). Matsusashi i wsp. uzyskali pod wpływem czteromiesięcznego leczenia sulindakiem (300 mg/dz) całkowitą lub też częściową remisję 13 spośród obserwowanych 20 gruczolaków jelita z obecnymi cechami atypii. Jednak badania te prowadzono bez grupy kontrolnej i wykluczono z nich osoby ze zmianami o dużym stopniu atypii (13).
Potwierdzenia wyników badań doświadczalnych i klinicznych szukano również poprzez szczegółową analizę danych epidemiologicznych. Już w 1988 r. zaobserwowano, że chorzy regularnie przyjmujący aspirynę rzadziej zapadają i umierają na raka okrężnicy (14). Kune i wsp. stwierdzili istnienie tej zależności analizując wiele czynników mogących potencjalnie wiązać się ze wzmożonym ryzykiem rozwoju raka jelita grubego. Zaskoczeni uzyskanymi wynikami, zbagatelizowali je uważając za przypadkowe i dopiero w 1991 roku kolejne publikacje potwierdzające doniesienia australijskich naukowców wywołały szersze zainteresowanie środowisk medycznych (15, 16).
Gridley i wsp. prospektywnie przeanalizowali częstość występowania nowotworów układu pokarmowego w grupie 11 000 osób z reumatoidalnym zapaleniem stawów przyjmujących przewlekle NLPZ. Stwierdzili, że u tych chorych była ona znacząco niższa niż w średniej populacji. Szczególnie korzystne działanie ochronne NLPZ wykazywały w stosunku do ryzyka rozwoju raka żołądka i jelita grubego (17).
W kolejnych latach przeprowadzono kilkanaście badań kontrolowanych (18, 19, 20) oraz prospektywnych (21, 22). Wszystkie, poza jednym w którym badana populacja była znacząco starsza (23), potwierdziły profilaktyczne działanie aspiryny i innych NLPZ w chorobie nowotworowej jelita grubego. Do niektórych z tych prac w charakterze grupy kontrolnej włączono również osoby przewlekle leczone acetamiofenem (paracetamolem). Nie stwierdzono jednak istnienia jakiejkolwiek zależności pomiędzy faktem długotrwałego przyjmowania paracetamolu, a ryzykiem rozwoju choroby nowotworowej.
W cytowanych doniesieniach wykazano, że regularne przyjmowanie aspiryny wiązało się ze średnio o połowę mniejszą zapadalnością na raka jelita grubego, przy czym obniżenie względnego ryzyka wystąpienia choroby nowotworowej było zależne od dawki NLPZ i czasu stosowania leku (24, 25). Tak więc również badania epidemiologiczne potwierdziły wyniki uzyskane w pracach doświadczalnych i klinicznych.
Warto natomiast podkreślić, że przedstawione korzystne efekty działania aspiryny nie dotyczą jej niższych dawek stosowanych w profilaktyce zawału serca (26), co więcej szczególnie silne działanie ochronne tego leku stwierdzane jest w przypadku jego przyjmowania przez okres ponad dziesięciu lat (24).
Znakomita większość przytoczonych badań koncentruje się na przeciwnowotworowym działaniu NLPZ w różnych modelach raka jelita grubego. Jednak podobne wyniki uzyskano również w przypadku nowotworów innych narządów – raka sutka (27, 32), pęcherza moczowego (28), przewodu pokarmowego (29), skóry (30) oraz płuc (31, 32). I tak, Schreinemachers i wsp. obserwowali znaczące zmniejszenie zapadalności na raka płuc u osób stosujących aspirynę (32), a Castonaguay i wsp. podając sulindak myszom przez okres 7 tygodni uzyskali w około 50% zahamowanie rozwoju raka tego narządu indukowanego u zwierząt chemicznie nitrozaminą, a więc karcinogenem obecnym w dymie papierosowym (31).
Mechanizm przeciwnowotworowego działania niesteroidowych leków przeciwzapalnych nie jest jasny (tab. 1).
Tabela 1. Mechanizmy działania niesteroidowych leków przeciwzapalnych na poziomie molekularnym i komórkowym, które mogą potencjalnie odpowiadać za ich efekt przeciwnowotworowy (Shiff 1999). (MAP – mitogen-activated protein, PPAR – peroxisomal proliferator-activated receptors).
| Mechanizmy zależne
od hamowania cyklooksygenazy | Mechanizmy niezależne
od hamowania cyklooksygenazy | Mechanizmy o nieznanej zależności
od hamowania cyklooksygenazy |
| Obrót tkankowy (proliferacja/apoptoza) | Obrót tkankowy (proliferacja/apoptoza) | Odpowiedź przeciwnowotworowa |
| Powstawanie karcinogenów | Transformacja komórkowa | Apoptoza (aktywacja onkogenu Myc) |
| Angiogeneza | Angiogeneza | Pobudzenie PPAR |
| | Naprawa DNA | |
| Apoptoza (indukcja kinaz MAP, blokowanie aktywacji czynnika jądrowego NF-kB) |
Z całą pewnością wiadomo natomiast, że za ich działanie przeciwbólowe, przeciwgorączkowe oraz przeciwzapalne odpowiada powinowactwo do cyklooksygenazy (COX) prowadzące do zahamowania aktywności katalitycznej tego enzymu i produkcji prekursorów mediatorów prozapalnych – prostaglandyn i tromboksanów (Ryc. 1). Aktualnie znane są dwie izoformy COX: COX-1 konstytutywna, stale obecna w wielu typach komórek, warunkująca różnorodne procesy fizjologiczne – m.in. proliferację i różnicowanie się komórek, oraz COX-2 indukowana postać enzymu, której ekspresja jest wzbudzana przez cytokiny, mediatory prozapalne i czynniki wzrostowe, odpowiadająca przede wszystkim za rozwój reakcji zapalnej. Obie postaci enzymu podlegają więc niezależnej regulacji i pełnią odmienne funkcje w ustroju. Uważa się obecnie, że hamowanie aktywności COX-2 warunkuje aktywność przeciwzapalną NLPZ, natomiast działanie na COX-1 odpowiada przede wszystkim za rozwój ich działań niepożądanych.
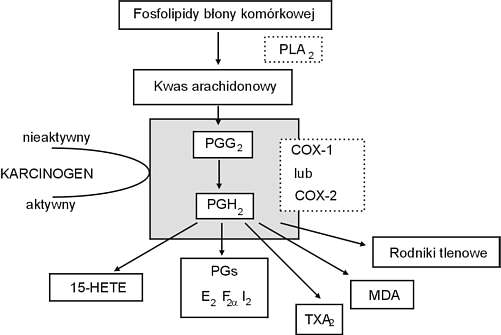
Ryc. 1. Rola izoenzymów cyklooksygenazy (COX) w procesie metabolizmu kwasu arachidonowego (PLA2 – fosfolipaza A2, PG – prostaglandyna, TXA – tromboksan, MDA – dwualdehyd malonowy).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Pollard M. et al.: The suppressive effect of piroxicam on autochthonous intestitial tumours in the rat. Cancer Lett., 1983, 21, 57-61. 2. Reddy B.B. et al.: Dose-related inhibition of colon carcinogenesis by dietary piroxicam, a nonsteroidal antiinflammatory drug, during different stages of rat colon tumour development. Cancer Res., 1987, 47:5340-6. 3. Rao C.V. et al.: Chemoprevention of colon carcinogenesis by sulindac, a nonsteroidal antiinflammatory agent. Cancer Res., 1995, 55:1464-72. 4. Craven P.A., DeRubertis F.R.: Effect of aspirin on 1,2-dimethylhydrazine-induced colonic carcinogenesis. Carcinogenesis, 1992, 13:541-6. 5. Hial V. et al.: Alternation of tumour growth by aspirin and indomethacin; studies with two transplantable tumours in mouse. Eur. J. Pharmacol., 1976, 37:367-76. 6. Ross D.S. et al.: Piroxicam inhibits growth of an adenocarcinoma isograft in Fischer rats. J. Surg. Res., 1988, 45:248-53. 7. Moorghen M. et al.: A protective effect of sulindac against chemically-induced primary colonic tumours in mice. J. Pathol., 1988, 156:341-47. 8. Waddell W.R., Loughry R.W.: Sulindac for polyposis of the colon. J. Surg. Oncol., 183, 24:83-7. 9. Waddell W.R. et al.: Sulindac for polyposis of the colon. Am. J. Surg., 1989, 157:175-9. 10. Labayle D. et al.: Sulindac causes regression of rectal polyps in familial adenomatous polyposis. Gastroenterology, 1991, 101:635-39. 11. Giardello F.M. et al.: Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N. Engl. J. Med., 1993, 328:1313-16. 12. Ladenheim J. et al.: Effect of sulindac on sporadic colonic polyps. Gastroenterology, 1995, 108:1083-7. 13. Matsuhashi N. et al.: Effect of sulindac on sporadic colorectal adenomatous polyps. Gut 1997, 40:344-9. 14. Kune G.A. et al.: Colorectal cancer risk, chronic illnesses, operations and medications: case control results from the Melbourne Colorectal Cancer study. Cancer res., 1988, 48:4399-4404. 15. Rosenberg L. et al.: A hypothesis: non-steroidal anti-inflammatory drugs reduce the incidence of large-bowel cancer. J. Natl. Cancer Inst., 1991, 83:355-8. 16. Thun M.J. et al.: Aspirin use and reduced risk of fatal colon cancer. N. Engl. J. Med., 1991, 325:1593-6. 17. Gridley G. et al.: Incidence of cancer among patients with rheumatoid arthritis. J. Natl. Canc. Inst., 1993, 85:307-10. 18. La Vecchia C. et al.: Aspirin and colorectal cancer. Br. J. Cancer, 1997, 76:675-7. 19. Suh O. et al.: Aspirin use, cancer, and polyps of the large bowel. Cancer 1993, 72:1171-7. 20. Freedman A.N. et al.: Aspirin use and p53 expression in colorectal cancer. Cancer Detect. Prev., 1998, 22:213-8. 21. Steering committee of the Physicians´ Health Study research group. Final report on the aspirine component of the ongoing Physicians´ Health Study. N. Engl. J. Med., 1989, 321:129-35. 22. Giovannuci E. et al.: Aspirin and the risk of colorectal cancer in woman. N. Engl. J. Med., 1995, 333:609-14. 23. Paganini-Hill A. et al.: Aspirin use and chronic diseases: a cohort study of the elderly. Br. Med. J., 1989, 299:1247-50. 24. Giovannuci E.: The prevention of colorectal cancer by aspirin use. Biomed. Pharmacother., 1999, 53:303-8. 25. Taketo M.M.: Cyclooxygenase-2 inhibitors in tumorigenesis (part II). J. Natl. Cancer Inst., 1998, 90:1609-20. 26. Gann P.H. et al.: Low-dose aspirin and incidence of colorectal tumours in the randomized trial. J. Natl. Cancer Inst., 1993, 85:1220-24. 27. McCormick D.L. et al.: Modulation of rat mammary carcinogenesis by indomethacin. Cancer Res., 1985, 45:1803-5. 28. Moon R.C. et al.: Chemoprevention of OH-BBN-induced bladder cancer in mice by piroxicam. Carcinogenesis 1993, 14:1487-9. 29. Rubio C.A.: Anti-tumour activity of indomethacin on experimental esophagal tumours. J. Natl. Cancer Inst., 1984, 72:702-5. 30. Fischer S.M. et al.: Inhibition of mouse skin tumour promotion by several inhibitors of arachidonic acid and metabolism. Carcinogenesis 1982, 3:1243-5. 31. Castonaquay A., Rioux N.: Inhibition of lung tumourgenesis by sulindac: comparison of two experimental protocols. Carcinogenesis 1997, 18:491-6. 32. Schreinemachers D.M., Everson R.B.: Aspirin use and lung, colon and breast cancer incidence in a prospective study. Epidemiology 1994, 5:138-46. 33. Sano H. et al.: Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res., 1995, 55:3785-9. 34. Kargman S.L. et al.: Expression of prostaglandin G/H synthase-1 and -2 protein in human colon cancer. Cancer Res., 1995, 55:2556-9. 35. Hixson E.D. et al.: Inhibition of prostaglandin synthesis: potential for chemoprevention of human colon cancer. Cancer Bull., 1991, 43:561-8. 36. Shiff S.J., Rigas B.: The role of cyclooxygenase inhibition in the antineoplastic effects of nonsteroidal antiinflammatory drugs (NSAIDs). J. Exp. Med., 1999, 190:445-50. 37. Shiff S.J., Rigas B.: Nonsteroidal anti-inflammatory drugs and colorectal cancer: evolving concepts of their chemopreventive actions. Gastroenterology 1997, 113:1992-8. 38. Hixson L.J. et al.: NSAID effect on the sporadic colon polyps. Am. J. Gastroenterol., 1993, 88:1652-6. 39. Tsujii M. et al.: Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell 1998, 87:803-9. 40. Skopińska-Różewska E. et al.: Inhibition of angiogenesis by sulindac and its sulfone metabolite (FGN-1): a potential mechanism for their antineoplastic properties. Int. J. Tissue React., 1998, 20:85-9. 41. Zhang X. et al.: Malignant transformation and antineoplastic actions of nonsteroidal antiinflammatory drugs (NSAIDs) on cyclooxygenase-null embryo fibroblasts. J. Exp. Med., 1999, 190:451-9. 42. Piazza G. et al.: Cyclic GMP (CG) phosphodiesterase (PDE) inhibition; a novel mechanism for the neoplastic properties of exisulind. Gastroenterology 1999, 116:A485. 43. Chan T.A. et al.: Mechanisms underlying nonsteroidal antiinflammatory drug-mediated apoptosis. Proc. Natl. Acad. Sci. USA, 1998, 95:681-6. 44. Herrmann C. et al.: Sulindac sulfide inhibits ras signaling. Oncogene 1998, 17:1769-76. 45. Ruschoff J. et al.: Aspirin suppresses the mutator phenotype associated with hereditary nonpolyposis colorectal cancer by genetic selection. Proc. Natl. Acad. Sci. USA, 1998, 95:11301-6. 46. Arvind P. et al.: PGE2 down-regulates the expression of HLA-DR antigen in human colon adenocarcinoma cell lines. Biochemistry 1995, 34:5604-9.

