© Borgis - Nowa Pediatria 2/2009, s. 50-55
*Małgorzata Mizerska-Wasiak
Nefropatia IgA
IgA nephropathy
Katedra i Klinika Pediatrii i Nefrologii Warszawskiego Uniwersytetu Medycznego
Kierownik Kliniki: prof. dr hab. med. Maria Roszkowska-Blaim
Streszczenie
IgA nephropathy (IgAN) is the most common type of glomerulonephritis worldwide. It is currently viewed as various clinical manifestations of renal lesions such as: gross/microscopic hematuria, proteinuria, nephrotic/nephritic syndrome or acute renal failure. IgA nephropathy has been reported in the past as a benign renal disease. Today we know that it may progress to chronic renal failure. Nephropathy in patients with IgAN progress already in childhood, leading to chronic renal failure in 5-30% of cases within 10 years and 25-50% of cases within 20 years. Complete remission has been reported in 3-30% patients with IgAN. The variants of initial manifestation and long-term follow up makes it difficult to asses the risk of progression of nephropathy and development of CRF in an individual patient. The treatment of IgAN is currently remains unstandardized among various centers worldwide.
The publication contains new data relative prevalence, pathogenesis, clinical features, risk factor of progression and treatment of IgA nephropathy in children.

Wstęp
Nefropatia IgA (IgA nephropathy, IgAN) uważana jest za najczęściej występujący na świecie typ kłębuszkowych zapaleń nerek (1, 2, 3, 4, 5, 6).
Nefropatia IgA została po raz pierwszy opisana przez Bergera w 1968 roku i nazwana od nazwiska autora chorobą Bergera (7). Przebiega pod postacią przewlekłego kłębuszkowego zapalenia nerek, z charakterystyczną obecnością depozytów IgA w mezangium (w przewadze nad innymi złogami) w badaniu biopsyjnym nerki w mikroskopie immunofluorescencyjnym (8, 9, 10).
Przez wiele lat nefropatia IgA była uważana za łagodną chorobę o dobrym rokowaniu (11) jednak odległe obserwacje wykazały, że choroba może prowadzić do przewlekłej niewydolności nerek (PNN). Progresja nefropatii zaczyna się już w dzieciństwie, a przewlekła niewydolność nerek występuje po 10 latach trwania choroby w 5-30% przypadków, po 20 latach odsetek ten wzrasta do 25-50% (2, 3, 4). Całkowitą remisję choroby opisano w IgAN w 3-30 % przypadków (2, 3).
Występowanie
Częstość występowania IgAN szacuje się na 18-40% wszystkich pierwotnych glomerulopatii w Japonii, Francji, Włoszech i Australii. Szczególnie często nefropatia IgA występuje w krajach Azji (4). W Japonii, z uwagi na wysoką częstość zachorowań, wśród dzieci między 6 a 18 r.ż. przeprowadzane jest scriningowe badanie moczu, mające na celu wczesne wykrycie objawów choroby (4). W Europie nefropatia IgA stanowi 10-30% glomerulopatii (12). We Włoszech rozpoznanie IgAN stanowi 30% w biopsjach wykonanych z powodu krwinkomoczu i 35% wśród biopsji wykonanych z powodu białkomoczu i krwinkomoczu (2, 3). IgAN rzadko występuje u rasy czarnej, w USA, Wielkiej Brytanii i Kanadzie tylko u 2-10% chorych (12).
Częstość występowania nefropatii IgA w Polsce jest zbliżona do średniej europejskiej; u pacjentów dorosłych na terenie Pomorza Gdańskiego w ostatnim dziesięcioleciu stanowi 19,5% wszystkich rozpoznań przewlekłych kłębuszkowych zapaleń nerek (13).
Etiologia i patogeneza
Nefropatia IgA jest pierwotnie idiopatyczną glomerulopatią, choć złogi IgA można stwierdzić w biopsji nerki także w przebiegu innych jednostek chorobowych. Wtórnie depozyty IgA mogą występować w nerkach m.in. w: celiakii, chorobach wątroby, cukrzycy, reumatoidalnym zapaleniu stawów, nieswoistych chorobach zapalnych jelit, w przebiegu HIV, HBV, chłoniaków, dermatitis herpetiformis i zespołów paraneoplastycznych w przebiegu nowotworów płuc i jelita grubego (13).
IgAN jest uważana za chorobę kompleksów immunologicznych (4). U 50-70% pacjentów dorosłych i 8-16% dzieci stwierdza się podwyższony poziom IgA w surowicy krwi (3, 4). Z badań in vitro wiadomo, że limfocyty krwi obwodowej pacjentów z nefropatią IgA produkują więcej IgA niż limfocyty zdrowych (15).
IgA występuje w dwóch podklasach IgA1 i IgA2 (15). W surowicy krwi 90% IgA to IgA1, IgA2 znajduje się w błonie śluzowej przewodu pokarmowego i układu oddechowego. IgA jest wydzielana w przewodzie pokarmowym i układzie oddechowym w odpowiedzi na antygeny bakteryjne i wirusowe, istnieje więc związek między infekcjami tych układów a rozwojem nefropatii IgA (14). Zidentyfikowane antygeny specyficzne wywołujące powstanie depozytów mezangialnego IgA, to heterogenna grupa, do której zaliczane są m.in: HSV, EBV, adenowirus, Helicobacter pylori, antygeny białek mleka. Produkowana w odpowiedzi na te antygeny immunoglobulina A pobudza fagocytozę przez IgA Fc receptor CD89 i powoduje aktywację dopełniacza drogą alternatywną, a także – wg ostatnich danych – lektynową przez wiązanie z MBL (mannan – binding lectin), co ma znaczenie w patogenezie IgAN (16).
IgA może występować w postaci monomerycznej lub łączyć się w polimery (polimeryczna IgA, pIgA) przy pomocy białkowego łańcucha J. U ludzi zdrowych większość pIgA jest produkowana przez układ immunologiczny błon śluzowych, u pacjentów z nefropatią IgA stwierdza się zwiększoną produkcję pIgA1 w szpiku, a obniżenie jej wytwarzania przez błony śluzowe (3). Badania wykazały, że pIgA1 jest głównym składnikiem depozytów na terenie kłębuszków nerkowych (17). Ta wzmożona produkcja stwierdzana jest też w odległej obserwacji u pacjentów z utrzymującymi się zmianami w moczu, a obniża się w okresie klinicznej remisji (tj. wraz z ustąpieniem zmian w moczu).
U chorych z nefropatią IgA, IgA1 ma zwiększoną zdolność do łączenia się w większe konglomeraty z powodu nieprawidłowej galaktozylacji w regionie zawiasowym tzw. O-glikozylacji, której przypisuje się obecnie kluczową rolę w patogenezie IgAN.
U chorych z IgAN stwierdza się w limfocytach B defekt β1,3-galaktozylotranferazy odpowiedzialnej za przyłączenie galaktozy do IgA (18). W stanie zdrowia IgA1 jest katabolizowana w wątrobie poprzez łączenie z receptorem asialoglikoproteinowym (ASGPR) (18, 19), w wyniku O-glikozylacji wątrobowy klirens IgA1 jest znacznie zmniejszony (18).
Cząsteczki IgA1 z obniżoną zawartością galaktozy, tworzą kompleksy immunologiczne, które łatwiej łączą się na terenie mezangium z fibronektyną, lamininą i kolagenem IV, co wyzwala powstanie nieswoistego odczynu zapalnego, w tym aktywację C3 dopełniacza i prowadzi do rozwoju nefropatii (20).
Komórki mezangialne wykazują też aktywność receptora ASGPR oraz mają zdolność produkcji pIgA1 i C3 w IgAN (18). Proliferacja komórek mezangium w IgAN zachodzi również przez pobudzenie receptora transferynowego (TfR) który preferencyjnie wiąże pIgA1, kompeksy IgA1 i nieprawidłowo glikozylowaną IgA1 i wywołuje wydzielanie interleukiny 6 (IL-6) i TGF-β (transforming growth factor – β) (18).
W patogenezie IgAN stwierdza się również udział kompleksów immunologicznych IgA – IgG, których znamiennie wyższy poziom występuje u pacjentów z IgAN w porównaniu z chorymi z innymi kłębuszkowymi zapaleniami nerek (39). Krążące kompleksy immunologiczne wyizolowane z surowicy pacjentów z IgAN wywołują także proliferację hodowanych in vitro kultur komórek mezangialnych (21).
Najnowsze badania dowodzą, że u myszy z deficytem β-galaktozylotransferazy rozwija się choroba podobna do ludzkiej nefropatii IgA, co podkreśla znaczenie nieprawidłowej galaktozylacji IgA1 w patogenezie IgAN (22).
Polimeryczna forma IgA1 indukuje ekspresję genów układu renina-angiotensyna oraz TGF-β znamiennie bardziej niż postać monomeryczna, co jest dowodem jej udziału w rozwoju przewlekłego procesu zapalnego i włóknienia nerek. TGF-β pobudza proliferację komórek mezangium, a hamowanie jego wydzielania powoduje zmniejszenie akumulacji białek macierzy zewnątrzkomórkowej (23).
Komórki mezangium produkują płytkowy czynnik wzrostu (PDGF, Platelet-derived growth factor) oraz łańcuch B PDGF (B-chain PDGF), które pobudzają proliferację mezangium. W badaniach nad eksperymentalnym modelem kłębuszkowego zapalenia nerek, hamowanie wydzielania tych czynników prowadzi do zmniejszenia proliferacji mezangium (23).
W nefropatii IgA obserwuje się także obecność makrofagów CD68 w nacieku zapalnym śródmiąższu oraz wzrost ich lokalnej proliferacji w zwierzęcych modelach eksperymentalnych, która pełni dużą rolę w odpowiedzi immunologicznej, nasileniu procesu zapalnego w nerkach i uszkodzeniu cewkowo-śródmiąższowym (23).
W patogenezie IgAN ważną rolę przypisuje się też czynnikom genetycznym. Wykazano też związek antygenów zgodności tkankowej HLA B12, Bw35 i B37 oraz HLA- DR1 i DR4 z rozwojem nefropatii IgA (24, 25).
Z badań niemieckich wiadomo, iż 9,6% pacjentów z IgAN miało 1 lub więcej krewnych z kłębuszkowym zapaleniem nerek (24).
Opisywany jest również wpływ polimorfizmu genu kodującego enzym konwertujący angiotensynę (ACE) oraz systemu genów renina-angiotensyna, a także polimorfizm innych genów. Gen ACE znajduje się na chromosomie 17 i składa się z 26 egzonów i 25 intronów. Polimorfizm ACE dotyczy 16 intronu składającego się z 287 par zasad.
Najbardziej znaczący dla badań genetycznych w IgAN jest tzw. polimorfizm I/D. „I” oznacza insercję – obecność 16 intronu, co w końcowym efekcie zmniejsza aktywność ACE, „D” – to delecja czyli brak tego intronu prowadzący do zwiększenia aktywności ACE. Genotyp ma działanie modulujące aktywność ACE w tkankach i w osoczu, najwyższą wykazano u homozygot DD (24). Powstała przy udziale enzymu konwertującego angiotensynę (ACE) angiotensyna II nasila w komórkach docelowych (w tym m.in. w nerkach) włóknienie i przerost. Stymuluje tam produkcję kolagenu, powoduje wzrost stężenia TGFβ i PDGF w komórkach mezangium i nabłonka cewek proksymalnych (26).
Maruyama i wsp. twierdzą, iż częstość genotypu DD jest znacząco wyższa u pacjentów z pogarszającą się funkcją nerek i może być markerem złej prognozy w nefropatii IgA (26). Stwierdzono też, że pacjenci z genotypem DD i ID wykazują znamiennie wyższą proteinurię, szkliwienie i zrosty z torebką Bowmana w kłębuszkach oraz uszkodzenie cewkowo-śródmiąższowe w porównaniu z pacjentami, u których stwierdzono genotyp II. Uważa się, że polimorfizm I/D ma związek z procesem przewlekłego uszkodzenia nerek, a nie ma wpływu na fazę ostrą procesu.
Objawy kliniczne
Nefropatia IgA manifestuje się u około połowy pacjentów makroskopowym krwiomoczem, który może wystąpić po raz pierwszy w trakcie lub w kilka dni po infekcji górnych dróg oddechowych i nawracać w czasie kolejnych infekcji (3, 4). Duże badania nefropatii IgA u dzieci pokazują, że w Europie i USA makroskopowy krwiomocz występuje u ok. 80% pacjentów (16, 17). 60% pacjentów ma więcej niż jeden epizod makroskopowej hematurii, a u 40% mogą występować bezobjawowe nawroty (bez obecności makroskopowego krwiomoczu) (27). Incydenty makroskopowej hematurii są rzadsze u dorosłych niż u dzieci z IgAN. Emancipator i wsp., badający grupę z przewagą pacjentów dorosłych opisują obecność krwiomoczu u 43% chorych (27). W badaniach japońskich krwiomocz stwierdzano u 18-32% dorosłych pacjentów i u 26% dzieci (4).
Wśród objawów może też występować długoletni krwinkomocz i/lub proteinuria o różnym nasileniu lub ostry zespół nefrytyczny z nadciśnieniem tętniczym i/lub ostrą niewydolnością nerek, a także zespół nerczycowy.
W badaniach Yoshikawy i wsp. wśród dzieci japońskich, krwinkomocz i/lub bezobjawowy białkomocz stwierdzano u 62% pacjentów, krwiomocz u 26%, a zespół nerczycowy lub nefrytyczny u 12% badanych (4).
W badaniach własnych autorki najczęściej występującymi objawami klinicznymi nefropatii IgA u dzieci był bezobjawowy białkomocz i/lub krwinkomocz, który obserwowano u 70% pacjentów, krwiomocz stwierdzano u 40%, zespół nefrytyczny lub nerczycowy u 20% pacjentów.
W badaniach laboratoryjnych stwierdza się podwyższenie stężenia IgA w surowicy krwi u 30-50% pacjentów dorosłych i tylko u 8-16% dzieci z nefropatią IgA (3). W materiale własnym podwyższony poziom IgA stwierdzano u 50% dzieci z nefropatią IgA
Zmiany histopatologiczne
Rozpoznanie nefropatii IgA można postawić tylko na podstawie biopsji nerki. W badaniu immunofluorescencyjnym stwierdza się w kłębuszkach nerkowych charakterystyczne złogi IgA, co przedstawia rycina 1. Ze złogami IgA mogą odkładać się depozyty składowej C3 dopełniacza, które obserwuje się w badaniach japońskich u ok. 60% pacjentów. Złogom IgA mogą towarzyszyć depozyty IgG u 32% dzieci, IgM u 8%, a IgG i IgM u 11% pacjentów (28).
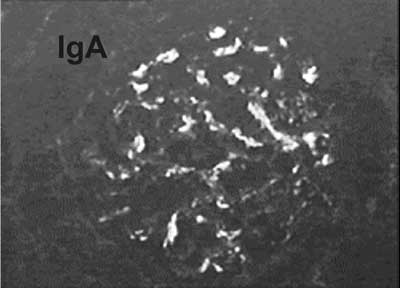
Ryc. 1. Kłębuszek nerkowy ze złogami IgA.
Charakterystyczną cechą nefropatii IgA w badaniu histopatologicznym jest też rozplem mezangium, w tym komórek i macierzy mezangialnej.
Zmiany stwierdzane w biopsji nerki w mikroskopie świetlnym są klasyfikowane wg Światowej Organizacji Zdrowia (WHO) w stopniach I-V, które oznaczają stopień zawansowania nefropatii (29):
I zmiany minimalne,
II rozplem mezangium <50% kłębuszków, nieliczne małe półksiężyce, bez zmian w tkance śródmiąższowej,
III rozlany rozplem mezangium, małe półksiężyce, ogniska przylegania pętli, ograniczony obrzęk i nacieki w tkance śródmiąższowej,
IV rozplem i szkliwienie we wszystkich kłębuszkach, przybytek komórek mezangium, w <50% kłębuszków półksiężyce i ogniska przylegania pętli naczyniowych do torebki, nacieki w tkance śródmiąższowej i zanik cewek,
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Wyatt RJ, Hogg RJ: Evidence-based assessment of treatment options for children with IgA nephropaties. Pediatr. Nephrol 2001; 16: 156-167. 2. Coppo R, D´Amico G: Factors predicting progression of IgA nephropaties J Nephrol 2005; 18: 503-512. 3. Coppo R: Pediatric IgA nephropathy: Clinical and Therapeutic perspectives Semin Nephrol 2008; 28 (1): 18-26. 4. Yoshikava N, Tanaka R, Iijima K: Pathophysiology and treatment of IgA nephropathy in children. Pediatr Nephrol 2001; 16: 446-457. 5. D´Amico G: Natural history of idiopathic IgA nephropathy: role of clinical and histological prognostic factors. Am J Kidney Dis 2000; 36: 227-237. 6. D´Amico G: Natural history of idiopatic IgA nephropathy and factors predictive of disease outcome. Semin Nephrol 2004; 24: 179-196. 7. Berger J, Hinglais N: Intercapillary deposits of IgA-IgG. J Urol Nephrol 1968; 74: 694-695. 8. Galla JH: Perspectives in clinical nephropathy. Kidney Int 1995; 47: 377-387. 9. Barrat MT, Avner ED, Harmon WE: Pediatric Nephrology. Lippincott Williams&Wilkins 1999; 691-707. 10. Berthoux FC, Mohey F, Afiani A: Natura history of primary IgA nephropathy. Semin Nephrol 2008; 28 (1): 4-9. 11. Yoshikava N, Iijima K, Ito H: IgA nephropathy in children. Nephron 1999; 83: 1-12. 12. Nolin L, Courteau M: Management of IgA nephropathy: evidence- based recommendation. Kidney Int 1999; 55 (Suppl 70): 56-62. 13. Książek A, Rutkowski B: Nefrologia. Wydawnictwo Czelej, Lublin 2004; 253-257. 14. Pouria S, Barratt J: Secondary IgA nephropathy. Semin Nephrol 2008; 28 (1): 27-37. 15. Floege J, Eitner F: Immune modulating therapy for IgA nephropathy: rationale and evidence. Semin Nephrol 2008; 28 (!): 38-47. 16. Ross A et al.: Glomerular activation of lectin pathway of complemet in IgA nephropathy is associated with more sever renal disease. J Am Soc Nephrol 2006; 17: 1724-1734. 17. Floege J, Feehally J: IgA nephropathy: recent developments. J Am Soc Nephrol 2000; 11: 2395-2403. 18. Moura IC et al.: Glycosylation and size of IgA1 are essential for interaction with mesangial transferring receptor in IgA nephropathy. J Am Soc Nephrol 2004; 16: 622-634. 19. Oortwijn BD, Ross A, Royle L: Differential Glycosylation of polymeric and monomeric IgA: A possible role in glomerular inflammation in IgA nephropathy. J Am Soc Nephrol 2006; 17: 3529-3539. 20. Van der Boog P et al.: Role of macromolecular IgA in IgA nephropathy. Kidney Int 2005; 67: 813-821. 21. Novak J et al.: IgA-containing immune complexes in IgA nephropathy differentially affect proliferation of mesangial cells. Kidney Int 2005; 67: 504-513. 22. Nishie T et al.: Development of immunoglobulin A nephropathy – like disease in β-1,4-galctosylotransferase-I-deficient mice. Am J Pathol 2007; 170: 447-456. 23. Lai KN et al.: Interaction between proximal tubular epithelial cells and infiltrating monocytes/T cells in the proteinuric state. Kidney Int 2007; 71 (6): 526-538. 24. Rambausek MH, Waldherr R, Ritz E: Immunogenetic findings in glomerulonephritis. Kidney Int 1993; 43: S3-S8. 25. Harden PN et al.: Polymorphisms in angiotensin-converting-enzym gen and progression of IgA nephropathy. Lancet 1995; 345: 1540-1542. 26. Maruyama K et al.: Polymorphisms of rennin-angiotensin system genes in childhood IgA nephropathy. Pediatr Nephrol 2001; 16 (4): 350-355. 27. Emancipator SN, Gallo GR, Lamm ME: IgA nephropathy: perspective on pathogenesis and classification. Clin Nephrol 1985; 24: 161-179. 28. Yoshikava N et al.: IgA nephropathy in children from Japan Child Nephrol Urol 1989; 9: 191-199. 29. Davison AM et al.: IgA nephropaties. Oxford textbook of clinical nephrology, Oxford University Press 1998; 537-570. 30. Andreoli SP, Bergstein JM: Treatment of sever IgA nephropathy in children. Pediatr Nephrol 1989; 3: 248-253. 31. Hass M, Rahman H, Cohn R: IgA nephropathy in children and adults: comparision of histologic features and clinical outcomes. NDT 2008; 23 (8): 2537-2545. 32. Ibels LS, Gyory AZ: IgA nephropathy: Analysis of natural history, important factors in the progression of renal disease and a review of the literature. Medicine (Baltimore) 1994; 73: 79-102. 33. Daniel L et al.: Tubular lesions determine prognosis of IgA nephropathy. Am J Kidney Dis 2000; 35: 13-20. 34. Nozawa R et al.: Clinicopathological features and the prognosis of IgA nephropathy in Japanese children in long-term observation. Clin Nephrol 2005; 64: 171-179. 35. Hastings MC, Delos Santos NM, Wyatt RJ: Renal survival in pediatric patients with IgA nephropathy. Pediatr Nephrol 2007; 22: 317-318. 36. Geddes CC et al.: A tricontinental view of IgA nephropathy. Nephrol. Dial Trnsplant 2003; 18: 1541-1548. 37. D´Amico G: Natural history of idiopatic IgA nephropathy and factors predictive of disease outcome. Semin Nephrol 2004; 24: 179-196. 38. Io H et al.: Relationship between levels of urinary type IV collagen ind renal injuries in patients with IgA nephropathy. J Clin Lab Analysis 2004; 18: 14-18. 39. Waldo BF et al.: Treatment of IgA nephropathy in children: efficacy of alternate-day oral prednison. Pediatr Nephrol 1993; 7: 529-532. 40. Kuriki M et al.: Steroid therapy reduces mesangial matrix accumulation in advanced IgA nephropathy. Nephrol Dial Transplant 2003; 18: 1311-1315. 41. Pozzi C et al.: Corticosteroids in IgA nephropathy: a randomised controlled trial. Lancet 1999; 353: 883-887. 42. Tamura S et al.: Corticosteroid therapy in patients with IgA nephropathy and impaired renal function. Clin Nephrol 2001; 55: 192-195. 43. Yoshikawa N et al.: A controlled trial of combined therapy for newly diagnosed severe childhood IgA nephropathy. J Am Soc Nephrol 1999; 10: 101-109. 44. Donadio JV Jr et al.: The long-term outcome of patients with IgA nephropathy treated with fish oil in a controlled trial. J Am Soc Nephrol 1999; 10: 1772-1777. 45. Tanaka H et al.: Combined therapy of enalapril and losartan attenuates histologic progression in immunoglobulin A nephropathy. Pediatr Int 2004; 46 (5): 576-579. 46. Roccatello D et al.: Report of intensive treatment of extracapillary glomerulonephritis with focus on crescentic IgA nephropathy. Nephrol Dial Transplant 1995; 10: 2054-2059. 47. Zhou WG et al.: Chronic tonsillitis and IgA nephropathy. Chin Med J 1993; 106: 770-772. 48. Van der Akker EH et al.: Long-term effects of pediatric adenotonsilectomy on serum immunoglobulins levels: results of a randomized control trial. Ann Allergy Asthma Immunol 2006; 97: 251-256. 49. Maes BD et al.: Mycofenolate mofetil in IgA nephropathy: Results of 3 year-prospective placebo-controlled randomized study. Kidney Int 2004; 65: 1842-1849. 50. Tang S et al.: Mycophenolate mofetil alleviates persistent proteinuria in IgA nephropathy. Kidney Int 2005; 68: 802-812. 51. Hogg RJ, Wyatt R: Scientific Planning Committee of the North American IgA Nephropathy Study. A randomized controlled trial of mycophenolate mofetil in patients with IgA nephropathy. BMC Nephrol 2004; 5, 3.