© Borgis - Postępy Nauk Medycznych 11/2007, s. 466-478
*Michał Pirożyński, Zbigniew Solarski
Astma oskrzelowa
Bronchial asthma
Klinika Anestezjologii i Intensywnej Terapii Centrum Medycznego Kształcenia Podyplomowego w Warszawie
Kierownik Kliniki: prof. dr hab. med. Jacek Jastrzębski
Streszczenie
Astma jest chorobą układu oddechowego znaną od dawna. Jest po alergicznym nieżycie nosa najczęstszą chorobą alergiczną. Autorzy przedstawiają prace nad patogenezą astmy, jej klasyfikacją i leczeniem.
Summary
Asthma is know for a long time. Following allergic rhinitis it is one of the most common allergic diseases of man. The authors discuss in this article the pathogenesis, classification and therapy of this disease.

Astma jest jednostką chorobową znaną od dość dawna. W starożytnej Grecji mianem astmy określano stan sapania, utrudnionego oddechu. Dopiero od około 30 lat astma znajduje się w środku uwagi nie tylko środowisk medycznych, mediów, ale również samych chorych. Dowodem tego są liczne publikacje naukowe, jak i popularnonaukowe. Astma jest uważana za jeden z głównych czynników wzrostu kosztów leczenia, jak również głównych i zasadniczych czynników rosnącego inwalidztwa wśród chorych. Nie dziwi, zatem zainteresowanie środowisk medycznych jej prewencją, patogenezą, epidemiologią, diagnostyką, jak i skuteczniejszą terapią. To rosnące zainteresowanie zaowocowało nie tylko liczbą prac na temat astmy, ale również zmieniało niemalże z dnia na dzień poglądy na jej patogenezę i leczenie (3).
Niewiele osób ma trudności ze zdefiniowaniem astmy oskrzelowej, twierdząc, że jest to schorzenie przebiegające z napadową dusznością, która ustępuje samoistnie lub pod wpływem leczenia. Oczywiście jest to definicja słuszna, lecz niepełna, nie mówi nam ani o przyczynach choroby ani o zjawiskach chorobowych zachodzących w układzie oddechowym pod jej wpływem. Dlatego w 1995 grupa ekspertów zaproponowała nowe określenie obejmujące nie tylko poprzednie jej elementy czynnościowe, ale również kliniczne (11). Przede wszystkim określiła elementy patogenezy astmy oskrzelowej. Obecnie astmę oskrzelową definiujemy w sposób następujący: „astma jest przewlekłą chorobą zapalną dróg oddechowych, w której uczestniczy wiele komórek i substancji przez nie uwalnianych. Przewlekłe zapalenie jest przyczyną nadreaktywności oskrzeli, prowadzącej do nawracających epizodów świszczącego oddechu, duszności, ściskania w klatce piersiowej i kaszlu, występujących szczególnie w nocy lub nad ranem. Epizodom tym zwykle towarzyszy rozlana obturacja oskrzeli o zmiennym nasileniu, często ustępująca samoistnie lub pod wpływem leczenia ”(3). Niestety żaden z przytoczonych elementów nie jest unikalny dla astmy. Zatem i ta definicja nie jest doskonała, nie obejmuje przecież tak charakterystycznej ciszy objawów klinicznych między kolejnymi napadami, jak również często prawidłowych wyników określających (6) przepływy powietrza przez drogi oddechowe (wskaźniki spirometryczne) u tych chorych (97). Nadreaktywność oskrzeli może nie być obecna w astmie sezonowej, w okresie między napadami, a może być obecna u chorych z nieżytem alergicznym nosa (18, 34). Nawet obecność eozynofilii, wydawałoby się tak charakterystycznej dla astmy, wcale nie jest cechą tak powszechnie występującą. Istnieje grupa chorych z klinicznymi jak i czynnościowymi objawami astmy, u których nie stwierdza się eozynofilowych nacieków błony śluzowej oskrzeli (19). Jak również istnieje grupa chorych z eozynofilowym zapaleniem błony śluzowej dróg oddechowych z przewlekłym kaszlem, reagujących na wziewne leki steroidowe, u których nie stwierdza się objawów obturacji (27). Należy również pamiętać o grupie chorych na astmę, u których stwierdza się w drzewie oskrzelowym nacieki neutrofilowe a nie eozynofilowe (101).
Epidemiologia i czynniki ryzyka
Wspomniane trudności w zdefiniowaniu astmy mają swój wpływ na badania epidemiologiczne nad nią. Badania długofalowe opierające się na historycznej definicji mogą nie odzwierciedlać prawdziwych danych epidemiologicznych. Również porównywanie grup chorych definiowanych różnymi określeniami, może prowadzić do błędnych wniosków (54).
Jedną z najmniej zmieniających się definicji były tzw. zgony astmatyczne. Dlatego też wszelkie spostrzeżenia dotyczące śmiertelnych powikłań astmy można przyjąć za wiarygodne. Badania retrospektywne wykazują stały powolny wzrost odsetka zgonów z powodu astmy w krajach cywilizowanych. Jednak gwałtowny przyrost zgonów nastąpił dwukrotnie, głównie w Nowej Zelandii, Australii, Wielkiej Brytanii i w Norwegii (90). W krajach tych nastąpił dwu- a nawet pięciokrotny ich wzrost w przeciągu 5 lat. Wydaje się, że spowodowane było to zastosowaniem nowego wówczas leku, nieselektywnego beta 2 mimetyku, izoproterenolu. Z chwilą jego wycofania nastąpił wyraźny spadek odsetka zgonów w tych krajach. Drugi szczyt zgonów z powodu astmy odnotowano w Nowej Zelandii. Ponownie połączono go z zastosowaniem wziewnego leku – fenoterolu. Jednak w tym wypadku wiadomo, że nie, fenoterol lecz zła kontrola astmy była główną przyczyną rosnącej śmiertelności. Z chwilą wprowadzenia wziewnych steroidów, przy jednoczesnym takim samym spożyciu fenoterolu nastąpił znaczny spadek odsetka zgonów (1). Niemniej badania wyraźnie wskazywały na rosnącą liczbę zgonów u chorych na astmę jak również na rosnącą częstość hospitalizacji chorych na astmę we wszystkich krajach zachodnich (68). Rosnący odsetek hospitalizacji wykazano w USA, gdzie przyrost w latach 1965-1984 wynosił 50% wśród dorosłych, a o ponad 200% wśród dzieci (12, 63).
Ten wzrost zapewne należy wiązać z rosnącą zapadalnością na astmę, jak również z rosnącą zapadalnością na choroby alergiczne (91). Trendy te odzwierciedla rosnąca częstość występowania dodatnich odczynów skórnych na alergeny inhalacyjne (7). Ciekawym zjawiskiem jest równoległy do wzrostu zapadalności na astmę wzrost zapadalności na alergiczny nieżyt nosa (4). Obserwacje te zostały niezależnie potwierdzone w Finlandii, Anglii oraz Szwecji (55, 81).
Wydaje się, że rzeczywiście mamy do czynienia ze wzrostem zachorowalności na astmę, ale również i na inne choroby alergiczne. Dotyczy to nie tylko krajów ucywilizowanych, ale również Trzeciego Świata (2).
Istnieją duże rozbieżności w częstości występowania objawów astmy oraz alergii w krajach a nawet w poszczególnych ośrodkach (2). Najwyższe współczynniki zapadalności notuje się w Anglii, Australii oraz Nowej Zelandii (20% populacji choruje na astmę), natomiast najniższe w Uzbekistanie, Grecji, Indonezji oraz krajach Europy Wschodniej (75).
Badania wykazały, że nie tylko atopia determinuje późniejszy rozwój astmy oskrzelowej oraz alergicznego nieżytu nosa, choć jej obecność jest jednym z najsilniejszych czynników ryzyka (74). Obecność atopii w rodzinie zwiększa ryzyko zachorowania na alergiczny nieżyt nosa pięciokrotnie a na astmę trzykrotnie (83).
Alergeny uczulające pochodzenia naturalnego stanowią niezależny czynnik ryzyka zachorowania na astmę oskrzelową. Wykazano to na przykładzie roztoczy kurzu domowego (79, 89). Inne alergeny, których obecność wiąże się ze zwiększonym ryzykiem zachorowania na astmę oskrzelową to alergeny kocie, karalusze oraz zarodniki Alternaria. Innym czynnikiem, który zwiększa ryzyko zachorowania na astmę jest stosowana dieta. Zależność tę wykazano w populacjach stosujących dietę typową dla krajów wysokorozwiniętych, o zwiększonym odsetku osób otyłych (38, 98). Ciekawe spostrzeżenia przyniosły dane z Nowej Zelandii wykazujące brak związku alergii na pyłki traw z zachorowaniem na astmę oskrzelową (87). Pozostałe czynniki słabiej powiązane z częstością zachorowania na astmę to: płeć męska, niska waga urodzeniowa, wcześniactwo, bierne palenie matki oraz wysoka konsumpcja soli w okresie ciąży (8, 55).
W ostatnim okresie w pracach badawczych obserwuje się duże zainteresowanie związkami częstych infekcji z występowaniem astmy oskrzelowej. Do niedawna były to badania oparte jedynie na obserwacji objawów klinicznych przeziębienia, poprzedzające zaostrzenia astmy lub wystąpienie tej jednostki chorobowej. Dodanie metod genetycznych do detekcji wirusów u chorych z objawami przeziębieniowymi, pozwoliło na wykazanie u prawie 80% dzieci i 40% dorosłych z zaostrzeniami astmy współistnienie zakażenia wirusowego (72). Wykazano również możliwość wywołania astmy przez powtarzające się zakażenia wirusowe, zwłaszcza te wywołane przez RSV (99). Sugestie te nie znalazły potwierdzenia w następnych latach. Badania przeprowadzone wśród dzieci sugerują możliwość ujawnienia, przez powtarzające się zakażenia wirusowe, predyspozycji do dominacji odpowiedzi Th2 zależnej, obecnej już w chwili zakażenia. Predyspozycja do odpowiedzi Th2 choć obecna, nie jest trwale ustalona. Przekierunkowanie jej w kierunku Th2, obecne w okresie płodowym, jest również przeniesione na okres neonatalny. Następnie ulega przeprogramowaniu na odpowiedź Th1. Jeżeli zjawisko to zostanie opóźnione, to niemowlę ma większe ryzyko w związku z przedłużoną odpowiedzią typu Th2 uczulenia na alergeny powietrznopochodne (96). Hipoteza ta sugeruje, że kontakt niemowlęcia z drobnoustrojami w obrębie dróg oddechowych jest odpowiedzialny na przekształcenie odpowiedzi Th2, fizjologicznie obecnej w okresie płodowym, w Th1 odpowiedzialnej za odporność skierowaną przeciwko drobnoustrojom. Zjawisko przesterowania Th1 – Th2 może być indukowane przez poszczególne wirusy. Niektóre z nich mają właściwości indukcji odpowiedzi Th2, odpowiedzi znacznie słabszej w eliminowaniu wirusów z ustroju (70).
„Chronienie” dziecka przed infekcjami wydaje się w świetle tej hipotezy odpowiedzialne za wzrost zachorowania na astmę wśród dzieci pochodzących z krajów wysokocywilizowanych (36). Badania epidemiologiczne wydają się potwierdzać tę hipotezę (95).
Dotychczas wydawało się, że zakażenia bakteryjne nie zaostrzają astmy oskrzelowej. Jednak znaczne zainteresowania budzą dwa gatunki bakterii Chlamydia i Mycoplasma. Oba jak wykazano występują znacznie częściej u chorych na astmę. W badaniu prospektywnym nad pozaszpitalnymi zapaleniami płuc wykazano, że u połowy chorych z zakażeniem Chlamydia pneumoniae występuje zjawisko skurczu oskrzeli (32). Podobne badania wykonane na populacji dzieci wykazało, że zakażenia Chlamydia pneumoniae może wywołać ostre napady świszczącego oddechu (23). Jeszcze ciekawsze dane przyniosły badania biopsyjne błony śluzowej oskrzeli chorych na astmę, w których wykazano u ponad połowy obecność w komórkach nabłonka oddechowego Mycoplasma pneumoniae (47).
Od dawna twierdzono, że istnieją predyspozycje genetyczne do występowania astmy oskrzelowej. Pierwsze prace pochodzą z 1860 roku, kiedy to Henryk Salter stwierdził, iż u 2 spośród 5 astmatyków można wykazać zjawisko dziedziczenia tej choroby. Te najwcześniejsze doniesienia są potwierdzane w najnowszych badaniach (86, 88).
Jak wspomniano, przewlekła astma oskrzelowa jest w swej istocie przewlekłym schorzeniem zapalnym, w którym dochodzi do naciekania ścian dróg oddechowych efektorowymi komórkami zapalnymi – limfocytami, makrofagami, mastocytami oraz granulocytami obojętno- i kwasochłonnymi. Mediatory zapalne wydzielane i syntetyzowane przez te komórki przyczyniają się do amplifikacji zapalenia w ścianach oskrzeli, jak również do powstawania zmian strukturalnych w drogach oddechowych (45, 46, 56, 66).
Zmiany strukturalne dróg oddechowych
W przebiegu astmy dochodzi do szeregu zmian strukturalnych w drogach oddechowych. Przede wszystkim dochodzi do złuszczania się komórek nabłonka. Cecha ta opisywana od lat, uważana była przez niektórych jako artefakt histologiczny powstały w trakcie opracowywania materiału. Jednak badania Jeffery´a wykazały niezbicie, że opisywane wcześniej złuszczanie całych płatów nabłonka oddechowego jest rzeczywiście charakterystyczne dla astmy oskrzelowej (43). Inną cechą charakterystyczną jest pojawienie się zmian metaplazji płaskonabłonkowej (20). Może ona współistnieć z poprzednio opisywanym złuszczaniem nabłonka. W ciężkiej astmie oskrzelowej hiperplazja komórek kubkowych jest jedną z bardziej charakterystycznych zmian. Przerost warstwy gruczołowej przyczynia się do wytwarzania nieprawidłowego gęstego śluzu tak charakterystycznego w tej postaci astmy (5).
Kolejną cechą astmy są nacieki eozynofilowe w ścianach oskrzeli (21, 51). Nacieki te składają się przede wszystkim z aktywowanych eozynofili. Ich intensywność i liczebność znacznie różni się między chorymi. Należy pamiętać, że u niektórych wręcz nie stwierdzono ich obecności (80).
Zmiany zachodzące w warstwie podnabłonkowej związane są z depozycją kolagenu typu III i V, fibronektyny i tenascyny (50). Źródłem tych protein są, mielofibroblasty, których populacja jest znacznie zwiększona w astmie (13).
W astmie w ścianach dróg oddechowych obserwuje się znaczny przyrost naczyń. Naczynia te pełnią znaczącą rolę w regulacji szerokości dróg oddechowych. Zwiększenie liczby naczyń, jak i wzrost przepuszczalności naczyń na skutek działania mediatorów zapalnych przyczynia się do powstawania obrzęku błony śluzowej, wpływając tym samym na szerokość światła dróg oddechowych (64).
Kolejnym czynnikiem wpływającym na drożność dróg oddechowych jest zachowanie się warstwy mięśni dróg oddechowych. W astmie spostrzegamy przerost oraz hipertrofię warstwy mięśniowej (15).
Czynnikiem zmieniającym sam kształt oskrzeli, decydującym o skuteczności terapii inhalacyjnej są zmiany w budowie chrząstek. Opisywane są przypadki zaburzeń ich budowy u chorych z zaawansowaną, ciężką astmą oskrzelową (33).
Przebudowa strukturalna dróg oddechowych wpływa również na fizjologię uwalnianego śluzu w drogach oddechowych. U większości chorych cechą charakterystyczną jest obecność gęstego, lepkiego śluzu tworzącego w ciężkiej astmie oskrzelowej czopy zatykające ujścia oskrzeli segmentarnych i płatowych. Należy pamiętać, że również zmiany w śluzie dotyczą nie tylko zmian ilościowych, ale również jakościowych. Zmiany w mikrokrążeniu, budowie nabłonka oddechowego wpływają na właściwości reologiczne śluzu. Nacieki z granulocytów kwasochłonnych przyczyniają się do pojawienia się kryształów Charkot – Leyden (zbudowanych z lisofosfolipazy eozynfolilowej) (85). Gęstość śluzu jest zwiększona na skutek wzrostu zawartości mucyn w jego składzie, co wpływa na pojawienie się charakterystycznych dla tej jednostki chorobowej spirali Curschmana (85). Złuszczony nabłonek wraz ze śluzem tworzą ciałka Creola (28). Wzrost przepuszczalności naczyń ścian oskrzeli, przyczyniający się do zwiększonego przesięku przyczynia się do wzrostu stężenia albuminy, DNA w wydzielinie oskrzelowej (25). Metaplazja i hiperplazja komórek kubkowych powodują zaleganie gęstego śluzu w najdrobniejszych oskrzelach (35). Wszystkie te zmiany powodują znaczne ograniczenie przepływu powietrza przez i tak już zwężone drogi oddechowe przyczyniając się do wzrostu duszności i coraz mniejszej tolerancji wysiłku u chorych na astmę oskrzelową.
Zmiany strukturalne dróg oddechowych przebiegają podobnie w dużych oskrzelach i w obwodowych drogach oddechowych. Nacieki kwasochłonne sięgają aż do pęcherzyków płucnych (48), a stopień odkładania kolagenu w okolicy pod błoną podstawną jest podobny w dużych jak i małych oskrzelach (43).
Choć od dawna istnieje podział na astmę atopową i nieatopową (określane na podstawie wyników testów skórnych oraz IgE) to zmiany zachodzące w drogach oddechowych w obu jednostkach nie różnią się zasadniczo między sobą (40, 57). Intensywność procesu chorobowego koreluje z liczbą komórek kwasochłonnych naciekających ściany oskrzeli jak również tych obecnych w wydzielinie oskrzelowej lub materiale z płukania oskrzelowo-pęcherzykowego (30). Natomiast ciężkość astmy nie jest już tak ewidentna w powiązaniu z włóknieniem ścian oskrzeli ( remodellingiem) (10, 16, 69, 82).
Zmiany zachodzące w ścianach dróg oddechowych, a zwłaszcza pojawienie się nacieków kwasochłonnych nie jest swoista dla astmy oskrzelowej. Nacieki takie spotyka się w przewlekłym zapaleniu oskrzeli, w przewlekłej obturacyjnej chorobie płuc i w innych zapalnych schorzeniach układu oddechowego, a także u chorych na alergiczny nieżyt nosa bez współistniejącej astmy oskrzelowej (26, 49). Również przerost warstwy mięśniowej nie jest spotykany tylko w astmie, ale również jest postrzegana w przewlekłym zapaleniu oskrzeli (76). Nacieki te różnicuje pojawienie się w astmie komórek CD4+, natomiast w pozostałych schorzeniach dominują limfocyty CD8+ (76) (ryc. 1).
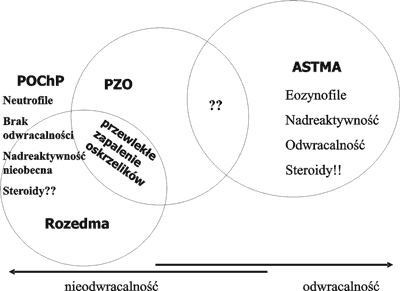
Ryc. 1. Cechy charakterystyczne astmy, POChP modyfikacja z pracy Barnesa (9).
Patofizjologia
Astma oskrzelowa jest schorzeniem, charakteryzującym się złożonymi mechanizmami patofizjologicznymi, wywołanymi interakcjami zachodzącymi między wieloma komórkami zapalnymi, a komórkami nabłonka oddechowego. Objawy kliniczne astmy – duszność, świszczący oddech, kaszel – odzwierciedlają złożoność procesów zachodzących w drogach oddechowych. Jak już wspomniano kluczowym elementem astmy jest proces zapalny zachodzący w oskrzelach. Proces ten to skutek interakcji wielu komórek ze sobą. Wiele prac badawczych miało na celu określenie komórki pełniącej rolę głównego dyrygenta całej reakcji zapalnej. Kandydatami były: mastocyt, eozynofile, komórka nabłonkowa dróg oddechowych, oraz limfocyt T CD4+. Obecnie oraz więcej dowodów przemawia za tezą, że to limfocyt T CD4+ jest tym dyrygentem (ryc. 2).
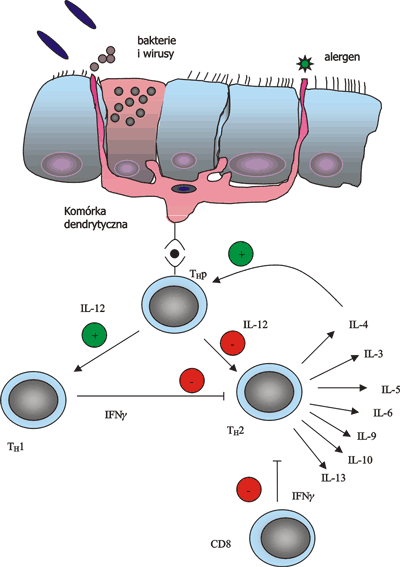
Ryc. 2. Schemat patomechanizmów zachodzących w astmie oskrzelowej.
W okresie ostatnich 10 lat udowodniono, że zjawiska immunologiczne zachodzące w ścianie oskrzeli są odpowiedzialne za objawy kliniczne astmy oskrzelowej. To alergiczne zapalenie rozpoczyna się z chwilą zetknięcia się alergenu powietrznopochodnego z komórkami prezentującymi antygen – komórkami dendrytycznymi i makrofagami. W zależności od rodzaju cytokin, które oddziaływują w tym momencie na limfocyt dziewiczy wytwarzane są różne klasy efektorowych limfocytów T (Th2, Th1) (ryc. 2). Źródłem tych różnic mogą być: zachowanie się samych limfocytów T, komórek prezentujących antygen, komórek nabłonka oddechowego, mastocytów, eozynofilii, makrofagów, a nawet miofibroblastów i komórek mięśni gładkich. Na ukierunkowanie odpowiedzi mają również wpływ współistniejące zakażenia wirusami i bakteriami, jak również charakter samych alergenów. Cytokiny wytwarzane przez komórki Th1 i Th2 różnią się. Tylko cytokiny Th2 (IL-3, IL-4, IL-5, IL-6, IL-9, IL-10 i IL-13) są związane z reakcjami zapalnymi charakterystycznymi dla astmy oskrzelowej. Cytokiny komórek Th1 wyhamowują reakcje zapalne alergiczne w drogach oddechowych (INF-γ i IL-12) (ryc. 2).
Spośród komórek prezentujących antygen – komórki nabłonka oddechowego, makrofagi, komórki dendrytyczne (zwane komórkami Langerhansa) – to te ostatnie wydają się najważniejsze. One to najsilniej indukują proces zapalny w drogach oddechowych. Znajdują się w jak i pod nabłonkiem oddechowym tak, aby jak najszybciej spełnić rolę komórki prezentującej antygen (52). Liczba ich jest podwyższona w astmie oskrzelowej. Mają one zdolność syntezowania, krytycznych dla komórek CD4+, IL-12, prostaglandyny E2 oraz IL-10.
Odpowiedź immunologiczną w astmie skrzelowej charakteryzuje obecność limfocytów CD4+ w nabłonku oddechowym jak również w warstwie podśluzówkowej (39). Komórki te kumulują się w płucach pod wpływem rozmaitych czynników, głównie IL-16. Wszystkie cytokiny są ważne w patogenezie astmy oskrzelowej jednak wydaje się, że dwie spośród nich pełnią najważniejszą rolę – IL-4 i IL-13. IL-4 jest odpowiedzialna za zmianę rodzaju produkowanych immunoglobulin przez limfocyty B z IgG1,IgG2,IgG3 i IgM w kierunku IgE i IgG4 oraz za zmianę fenotypu limfocytów Th0 w kierunku Th2. IL-13 może stymulować syntezę IgE, ale ważniejsza jej rola to mediacja nadreaktywności oskrzeli, hiperplazji komórek kubkowych i eozynofilii oskrzelowej (31, 94, 103).
Spośród pozostałych cytokin dość istotne są IL-12 i IL-10. IL-10 syntetyzowana jest nie tylko przez komórki Th2, ale również przez komórki nabłonka oddechowego i makrofagi. IL-10 reguluje alergiczne zapalenie dróg oddechowych poprzez swój wpływ na komórki dendrytyczne (17).
Nabłonek dróg oddechowych poza funkcją ochronną a także udziałem w mechanizmach obronnych (transport śluzowo-rzęskowy) pełni szereg dość istotnych funkcji. Komórki nabłonka uczestniczą w lokalnych reakcjach immunologicznych. Pod wpływem różnych bodźców posiadają zdolność wytwarzania pozapalnych mediatorów oraz protein. Efektem, czego jest modelowanie reakcji zapalnej na poziomie nabłonka (73, 77). Uwalniane czynniki mają różnorodny wpływ na organizm m.in. działają jako czynniki chemotaktyczne dla krążących leukocytów, regulują napięcie mięśni oskrzeli, regulują wydzielanie śluzu przez gruczoły w drogach oddechowych (37).
Makrofagi oprócz nieswoistych funkcji obronnych realizowanych między innymi poprzez zjawisko fagocytozy, uczestniczą w zapoczątkowaniu i podtrzymywaniu procesu zapalnego wywołanego przez alergen. Makrofagi pełnią aktywną rolę w swoistych procesach immunologicznych jako komórki przygotowujące antygen, ściśle współpracują z limfocytami, mają wpływ na chemotaksję i aktywność granulocytów obojętnochłonnych, a także zdolność uwalniania wielu substancji o właściwościach mediatorów zapalenia. Zaliczają się do nich wolne rodniki tlenowe, enzymy proteolityczne, antyproteazy, hormony polipeptydowe, lipidy bioaktywne (PAF, leukotrieny, prostaglandyny) (65, 93).
Makrofagi umiejscowione w obrębie miąższu płucnego można podzielić na makrofagi pęcherzyków płucnych, makrofagi dróg oddechowych, makrofagi tkanki śródmiąższowej i makrofagi w naczyniach włosowatych łożyska płucnego. Najlepiej poznane ze względu na łatwą technikę uzyskania za pomocą płukania oskrzelowo-pęcherzykowego, są makrofagi pęcherzyków płucnych.
Na powierzchni makrofagów znajdują się receptory umożliwiające im interakcję ze środowiskiem. Ze względu na właściwości można je podzielić na immunologiczne i nieimmunologiczne. Do immunologicznych zaliczają się receptory dla fragmentów Fc immunoglobulin, receptory dla składników dopełniacza, a także receptory głównego układu zgodności tkankowej klasy II. Obecność receptorów dla fragmentów Fc immunoglobulin, a także składnika dopełniacza C3b umożliwia makrofagom proces fagocytozy. Oprócz receptorów określanych jako immunologiczne, makrofagi posiadają także wiele receptorów nieimmunologicznych potwierdzających wielokierunkowy wpływ tych komórek na homeostazę organizmu.
Receptory układu zgodności tkankowej klasy II umożliwiają makrofagom prezentację antygenów limfocytom T. Limfocyty T swoiście wiążące epitopy alergenów uwalniają cytokiny, między innymi TNF-α, pobudzając w ten sposób inne funkcje makrofagów. Makrofagi odgrywają szczególnie dużą rolę w prezentacji antygenów pochodzących z dużych cząstek lub bakterii, czego nie potrafią inne komórki mające zdolność prezentacji antygenów, tj. limfocyty B czy komórki dendrytyczne.
Bardzo istotny jest wpływ makrofagów na granulocyty, a przede wszystkim na eozynofile poprzez wydzielane mediatory. Produkowany między innymi przez makrofagi GM-CSF stymuluje dojrzewanie i różnicowanie granulocytów, pobudza eozynofile do produkcji LTC4 i PAF, a także zwiększa uwalnianie histaminy z bazofilów. Współpracując z limfocytami, uczestniczą aktywnie w swoistej odpowiedzi immunologicznej. Aktywowane limfocyty T wydzielają natomiast IL-4, która między innymi posiada zdolność zwiększania ekspresji Fc-Σ-RII na makrofagach. Ponadto makrofagi wraz z płytkami i limfocytami syntetyzują i wydzielają grupę substancji określanych jako czynniki uwalniające histaminę. Komórki te mogą także bezpośrednio wpływać na kurczliwość mięśni gładkich poprzez uwalnianie prostanoidów.
Alergiczne reakcje typu I są bardzo istotne w zaostrzeniach astmy oskrzelowej. To one odpowiedzialne są za utrzymywanie się przewlekłego stanu zapalnego w drogach oddechowych. W reakcjach tych na skutek połączenia IgE z receptorem komórki tucznej lub bazofila dochodzi do uwalniania szeregu bardzo aktywnych substancji – histaminy, tryptazy, chymazy, leukotrienów, czynników płytkowych, oraz IL-4, IL-5, TNF-α. Wszystkie te czynniki są odpowiedzialne za nadmierne wydzielanie śluzu oraz zjawisko nadreaktywności oskrzeli, aktywację fibroblastów, degradację neuropeptydów, IgE zależną aktywację mastocytów i bazofili. Razem stanowią części składowe wczesnej i późnej reakcji astmatycznej na wdychany alergen (24).
Wśród komórek migrujących za istotne w rozwoju astmy uważa się krwinki kwasochłonne. Po aktywacji komórek znajdujących się w drogach oddechowych, zwłaszcza komórek tucznych, makrofagów oraz komórek nabłonkowych dochodzi do napływu eozynofili, jak również pozostałych komórek zapalnych granulocytów kwasochłonnych, zasadochłonnych, granulocytów obojętnochłonnych i płytek krwi.
Rola eozynofilów w reakcjach alergicznych w dalszym ciągu nie jest do końca wyjaśniona. Eozynofile powstają w szpiku, osiągają pełną dojrzałość w ciągu 5-6 dni. Za proliferację, zróżnicowanie eozynofilów odpowiedzialne są GM-CSF i IL-5, które mają wpływ na rozwój oraz przedłużanie żywotności eozynofilów. Liczba eozynofilów we krwi jest kilkusetkrotnie mniejsza niż w tkance łącznej. Szczególnie dużo tych komórek spotyka się w tkankach objętych procesem zapalnym o podłożu alergicznym (29).
Można wyróżnić dwie postacie eozynofilów hypodensyjne (lekkie) i normodensyjne, odzwierciedlające prawdopodobnie stan aktywacji tych komórek. Te pierwsze uznawane są za aktywowane, te drugie – za spoczynkowe. Lekkie eozynofile mają mniej ziaren w cytoplazmie, ale więcej receptorów dla składników dopełniacza i dla IgE. U zdrowych osób lekkie eozynofile stanowią ok. 10% eozynofilów, u astmatyków odsetek ten może sięgać 30-60% (100).
Mediatory wydzielone przez eozynofile można podzielić na 3 grupy: mediatory ziaren, mediatory błony komórkowej i mediatory produkowane i uwolnione podczas aktywacji. Są nimi wydzielane przez te komórki MBP (major basic protein), ECF (eosinophil cathionic protein), EDN (eosinophil derived neurotoxin) i EPO (eosinophil peroxidase) powodujące uszkodzenie i złuszczenie nabłonka oskrzeli oraz zatrzymanie ruchu rzęsek.
Od dawna uważa się, że liczba eozynofilów we krwi i w tkankach wzrasta w przebiegu astmy oskrzelowej. Za zjawisko to odpowiedzialne są różne czynniki, działające chemotaktycznie lub aktywująco. Można wśród nich wyróżnić: ECF-A wydzielany przez komórki tuczne ( eosinophil chemotactic factor of anaphylaxis), GM-CSF ( granulocyte- macrophage colony stimulating factor), IL-5, IL-2 i IL-3 wydzielane przez limfocyty, TNF wydzielany przez makrofagi.
W miejscu reakcji alergicznych eozynofile mogą swoiście uczestniczyć w toczącym się procesie zapalnym. Dzieje się tak dzięki obecności na ich powierzchni receptorów FcER II. FcER może indukować fagocytozę cząsteczek opłaszczonych przeciwciałami IgE, ale nie jest to prawdopodobnie jego główna funkcja. W wyniku proteolizy może być on uwolniony w formie rozpuszczalnej do środowiska. Sam FcER II ma właściwości autoproteolityczne. Podwyższony poziom IgE w środowisku zwiększa gęstość występowania FcER II na komórkach. Nie wynika to jednak ze zwiększonej syntezy tego receptora, lecz prawdopodobnie jest następstwem przyłączenia się IgE do Fc RII i hamowania proteolizy uwalniającej go do środowiska. FcER II bierze udział w aktywacji odpowiedzi humoralnej w zakresie przeciwciał IgE i prawdopodobnie aktywuje limfocyty B. Istnieją dwie odmiany tego receptora: FcERII i FcERI i b. Pierwszy występuje tylko na limfocytach B, a drugi na monocytach i eozynofilach oraz na limfocytach B pobudzonych przez IL-4. Różnią się one w odcinku cytoplazmatycznym.
Można założyć, że przy niewielkim narażeniu na alergen „przyciąganie” w miejscu objętym reakcją zapalną jest do pewnego stopnia przypadkowe i może mieć charakter korzystny. Eozynofile są w stanie fagocytować kompleksy immunologiczne, mogą inaktywować mediatory np. histaminę, leukotrieny. Natomiast w przypadku masywnego i długotrwałego narażenia na alergeny mogą stać się komórkami współuczestniczącymi w procesie destrukcji tkanek. Uszkodzenie tkanek w przebiegu astmy jest w dużej mierze związane z działaniem uwolnionych mediatorów.
Eozynofile mogą odgrywać w przebiegu astmy oskrzelowej rolę korzystną, obojętną lub szkodliwą. Zależy to nie tylko od czasu trwania narażenia na alergen, ale także od „jakości” bodźca, czego dowodem jest różne zachowanie się eozynofilii w zależności od sygnału aktywującego.
W ostatnich latach pojawiają się doniesienia na temat roli neutrofili w astmie oskrzelowej. Choć ich liczba w świetle dróg oddechowych oraz w ścianach dróg oddechowych nie jest podwyższona, to jednak istnieją populacje chorych na astmę, u których stwierdzamy podwyższony odsetek tych komórek. Zwłaszcza dotyczy to chorych, u których stwierdzamy astmę o bardzo ciężkim przebiegu (42, 100).
Drogi oddechowe są bogato unerwione. Wewnątrzścienny układ nerwowy stanowiący element układu autonomicznego składa się ze zwojów, komórek i włókien nerwowych. Receptory czuciowe generujące sygnały dla nerwów są określone zbiorczo jako receptory kaszlu. Wyróżnia się receptory włókien C, receptory wrażliwe na bodźce drażniące ( irritant receptors), receptory szybko adaptujące ( RAR – rapidly adapting receptors) i mięśniowe receptory wolno adaptujące ( SAR – slowly adapting receptors).
Oprócz generowania sygnałów aferentnych, autonomiczne zakończenia czuciowe w trakcie stymulacji wydzielają neuropeptydy NANC ( nonadrenergic noncholinergic), które wpływają na czynność mięśni gładkich oskrzeli, wytwarzanie śluzu przez gruczoły podśluzowe oraz transport jonów przez nabłonek. W ścianie dróg oddechowych wykazano istnienie tachykinin (NKA, NKB, SP), CGRP,VIP, PHM/L. Z zakończeń nerwowych może uwalniać się też tlenek azotu. Fizjologiczna rola wewnątrzściennego układu nerwowego polega na scalaniu czynności ściany drogi oddechowej w skuteczne reakcje oczyszczające i utrzymujące drożność.
W czasie przeciążenia mechanizmów fizjologicznych stymulacja układu NANC doprowadza do wystąpienia reakcji tkankowych, określanych jako neurogenne zapalenie. Dochodzi wtedy do lokalnego przekrwienia, zwiększenia przepuszczalności naczyń krwionośnych i nabłonka oraz do aktywacji komórek odpornościowych i uwalniania innych, typowych mediatorów reakcji zapalnych. Mechanizm odruchu aksonalnego może dodatkowo powodować skurcz błony mięśniowej.
W nabłonku znajdują się też komórki neuroendokrynowe. Są to komórki neuroepitelialne. Skupienia wewnątrzbłonkowe tych komórek są nazywane ciałami neuroepitelialnymi ( neuropithelial bodies). Komórki te zawierają neuromediatory, a czasem też i neuropeptydy (serotonina, histamina, CGRP i inne).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Fenoterol and asthma deaths. Drug. Ther. Bull., 1990 Dec. 10, 28, 25: 65-6.
2. Worldwide variation in prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and atopic eczema: ISAAC. The International Study of Asthma and Allergies in Childhood (ISAAC) Steering Committee. Lancet 1998; 25, 351, 9111: 1225-32.
3. Światowa strategia rozpoznawania, leczenia i prewencji astmy. Raport NHLBI/WHO. Med. Prakt., 2002, wydanie specjalne 6: 1-181.
4. Aberg N: Asthma and allergic rhinitis in Swedish conscripts. Clin. Exp. Allergy 1989; 19, 1: 59-63.
5. Aikawa T, et al.: Marked goblet cell hyperplasia with mucus accumulation in the airways of patients who died of severe acute asthma attack. Chest. 1992; 101, 4: 916-21.
6. Ayres JG: Classification and management of brittle asthma. Br. J. Hosp. Med., 1997; 16, 57, 8: 387-9.
7. Barbee RA: The epidemiology of asthma. Monogr. Allergy 1987; 21: 21-41.
8. Barbee RA, Murphy S: The natural history of asthma. J. Allergy Clin. Immunol., 1998; 102, 4 Pt. 2: 65-72.
9. Barnes PJ: Mechanisms in COPD: differences from asthma. Chest. 2000; 117, 2 Suppl: 10-4.
10. Boulet LP, et al.: Bronchial subepithelial fibrosis correlates with airway responsiveness to methacholine. Chest. 1997; 112, 1: 45-52.
11. Bousquet J, et al.: GINA guidelines on asthma and beyond. Allergy 2007; 62, 2: 102-12.
12. Braman SS: The global burden of asthma. Chest. 2006 Jul.,130, 1 Suppl: 4-12.
13. Brewster CE, et al.: Myofibroblasts and subepithelial fibrosis in bronchial asthma. Am. J. Respir. Cell. Mol. Biol., 1990; 3, 5: 507-11.
14. Brown PJ, et al.: Asthma and irreversible airflow obstruction. Thorax 1984; 39, 2: 131-6.
15. Carroll N, et al.: The structure of large and small airways in nonfatal and fatal asthma. Am. Rev. Respir. Dis., 1993; 147, 2: 405-10.
16. Chetta A, et al.: Airways remodeling is a distinctive feature of asthma and is related to severity of disease. Chest. 1997; 111, 4: 852-7.
17. Chung F: Anti-inflammatory cytokines in asthma and allergy: interleukin-10, interleukin-12, interferon-gamma. Mediators. Inflamm., 2001; 10, 2: 51-9.
18. Cockcroft DW, et al.: Bronchial reactivity to inhaled histamine: a method and clinical survey. Clin. Allergy 1977; 7, 3: 235-243.
19. Crimi E, et al.: Dissociation between airway inflammation and airway hyperresponsiveness in allergic asthma. Am. J. Respir. Crit. Care Med., 1998; 157, 1:4-9.
20. Cutz E, et al.: Ultrastructure of airways in children with asthma. Histopathology 1978; 2, 6: 407-21.
21. Djukanovic R, et al.: Quantitation of mast cells and eosinophils in the bronchial mucosa of symptomatic atopic asthmatics and healthy control subjects using immunohistochemistry. Am. Rev. Respir. Dis., 1990; 142, 4: 863-71.
22. Elliott MW: The language of breathlessness. Monaldi Arch. Chest. Dis., 1998; 53, 6: 664-8.
23. Emre U, et al.: The association of Chlamydia pneumoniae infection and reactive airway disease in children. Arch. Pediatr. Adolesc. Med., 1994; 148, 7: 727-32.
24. Fahy JV, et al.: The effect of an anti-IgE monoclonal antibody on the early – and late-phase responses to allergen inhalation in asthmatic subjects. Am. J. Respir. Crit. Care Med., 1997; 155, 6: 1828-34.
25. Fahy JV, et al.: Cellular and biochemical analysis of induced sputum from asthmatic and from healthy subjects. Am. Rev. Respir. Dis., 1993; 147, 5: 1126-31.
26. Foresi A, et al.: Eosinophils, mast cells, and basophils in induced sputum from patients with seasonal allergic rhinitis and perennial asthma: relationship to methacholine responsiveness. J. Allergy Clin. Immunol., 1997; 100, 1: 58-64.
27. Gibson PG, et al.: Chronic cough with eosinophilic bronchitis: examination for variable airflow obstruction and response to corticosteroid. Clin. Exp. Allergy 1995; 25, 2: 127-32.
28. Gleich GJ: The pathology of asthma: with emphasis on the role of the eosinophil. N. Engl. Reg. Allergy Proc., 1986; 7, 5: 421-4.
29. Gronke L, et al.: The relationship between airway hyper-responsiveness, markers of inflammation and lung function depends on the duration of the asthmatic disease. Clin. Exp. Allergy 2002; 32, 1: 57-63.
30. Grootendorst DC, et al.: Comparison of inflammatory cell counts in asthma: induced sputum vs bronchoalveolar lavage and bronchial biopsies. Clin. Exp. Allergy 1997; 27, 7: 769-79.
31. Grunig G, et al.: Requirement for IL-13 independently of IL-4 in experimental asthma. Science 1998; 18, 282, 5397: 2261-3.
32. Hahn DL, et al.: Association of Chlamydia pneumoniae (strain TWAR) infection with wheezing, asthmatic bronchitis, and adult-onset asthma. JAMA 1991; 10, 266, 2: 225-30.
33. Haraguchi M, et al.: Morphometric analysis of bronchial cartilage in chronic obstructive pulmonary disease and bronchial asthma. Am. J. Respir. Crit. Care Med., 1999; 159, 3: 1005-13.
34. Hensley MJ, et al.: Seasonal variation in non-specific bronchial reactivity: a study of wheat workers with a history of wheat associated asthma. Thorax 1988; 43, 2: 103-7.
35. Hogg JC: The pathology of asthma. Clin. Chest. Med., 1984 ; 5, 4: 567-71.
36. Holt PG: Environmental antigens and atopic disease: underlying mechanisms and prospects for therapy and prophylaxis. Mol. Med. Today 1995; 1, 6: 292-8.
37. Holtzman MJ, et al.: Immunity, inflammation, and remodeling in the airway epithelial barrier: epithelial-viral-allergic paradigm. Physiol. Rev., 2002; 82, 1: 19-46.
38. Huang SL, et al.: Association between body mass index and allergy in teenage girls in Taiwan. Clin. Exp. Allergy 1999; 29, 3: 323-9.
39. Humbert M, et al.: Elevated expression of messenger ribonucleic acid encoding IL-13 in the bronchial mucosa of atopic and nonatopic subjects with asthma. J. Allergy Clin. Immunol., 1997; May, 99, 5: 657-65.
40. Humbert M, et al.: IL-4 and IL-5 mRNA and protein in bronchial biopsies from patients with atopic and nonatopic asthma: evidence against „intrinsic” asthma being a distinct immunopathologic entity. Am. J. Respir. Crit. Care Med., 1996; 154, 5: 1497-504.
41. Irwin RS, et al.: Chronic cough. The spectrum and frequency of causes, key components of the diagnostic evaluation, and outcome of specific therapy. Am. Rev. Respir. Dis., 1990; 141, 3: 640-7.
42. Jeffery PK: Lymphocytes, chronic bronchitis and chronic obstructive pulmonary disease. Novartis. Found. Symp., 2001, 234: 149-61.
43. Jeffery PK, et al.: Bronchial biopsies in asthma. An ultrastructural, quantitative study and correlation with hyperreactivity. Am. Rev. Respir. Dis., 1989; 140, 6: 1745-53.
44. Joos G, et al.: The respiratory effects of neuropeptides. Eur. J. Respir. Dis. Suppl., 1986, 144: 107-36.
45. Kay AB: Natural killer T cells and asthma. N. Engl. J. Med., 2006; 16, 354, 11: 1186-8.
46. Kay AB: The role of T lymphocytes in asthma. Chem. Immunol. Allergy 2006, 91: 59-75.
47. Kraft M, et al.: Detection of Mycoplasma pneumoniae in the airways of adults with chronic asthma. Am. J. Respir. Crit. Care Med., 1998; 158, 3: 998-1001.
48. Kraft M, et al.: Alveolar tissue inflammation in asthma. Am. J. Respir. Crit. Care Med., 1996; 154, 5: 1505-10.
49. Lacoste JY, et al.: Eosinophilic and neutrophilic inflammation in asthma, chronic bronchitis, and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol., 1993; 92, 4: 537-48.
50. Laitinen A, et al.: Tenascin is increased in airway basement membrane of asthmatics and decreased by an inhaled steroid. Am. J. Respir. Crit. Care Med., 1997; 156, 3 Pt. 1: 951-8.
51. Laitinen LA, et al.: Airway mucosal inflammation even in patients with newly diagnosed asthma. Am. Rev. Respir. Dis., 1993; 147, 3: 697-704.
52. Lambrecht BN, et al.: Dendritic cells as regulators of the immune response to inhaled allergen: recent findings in animal models of asthma. Int. Arch. Allergy Immunol., 2001; 124, 4: 432-46.
53. Li JT, O´Connell EJ: Clinical evaluation of asthma. Ann. Allergy Asthma Immunol., 1996; 76, 1: 1-13.
54. Lugogo NL, Kraft M: Epidemiology of asthma. Clin. Chest Med., 2006; 27, 1: 1-15, v.
55. Lundback B: Epidemiology of rhinitis and asthma. Clin. Exp. Allergy 1998; 28 Suppl. 2: 3-10.
56. Mathur SK, Busse WW: Asthma: diagnosis and management. Med. Clin. North Am., 2006; 90, 1: 39-60.
57. Mattoli S, et al.: Cellular and biochemical characteristics of bronchoalveolar lavage fluid in symptomatic nonallergic asthma. J. Allergy Clin. Immunol., 1991; 87, 4: 794-802.
58. McFadden ER Jr.: Exertional dyspnea and cough as preludes to acute attacks of bronchial asthma. N. Engl. J. Med., 1975; 13, 292, 11: 555-9.
59. McFadden ER Jr, Gilbert IA: Asthma. N. Engl. J. Med., 1992; 31, 327, 27: 1928-37.
60. McFadden ER Jr, Hejal R: Asthma. Lancet 1995; 13, 345, 8959: 1215-20.
61. McFadden ER Jr, et al.: Acute bronchial asthma. Relations between clinical and physiologic manifestations. N. Engl. J. Med. 1973; 1, 288, 5: 221-5.
62. Merkus PJ, de Jongste JC: Inhaled corticosteroids and long term outcome in adults with asthma. Thorax 2006; 61, 11: 1011.
63. Mitchell EA: International trends in hospital admission rates for asthma. Arch. Dis. Child., 1985; 60, 4: 376-8.
64. Mitzner W, et al.: Is asthma a vascular disorder? Chest. 1995; 107, 3 Suppl: 97-102.
65. Nathan CF: Secretory products of macrophages. J. Clin. Invest., 1987; 79, 2: 319-26.
66. Nigo YI, et al.: Regulation of allergic airway inflammation through Toll-like receptor 4-mediated modification of mast cell function. Proc. Natl. Acad. Sci., USA 2006; 14, 103, 7: 2286-91.
67. O´Byrne PM, Parameswaran K: Pharmacological management of mild or moderate persistent asthma. Lancet 2006; 26, 368, 9537: 794-803.
68. Oates JA, et al.: Clinical implications of prostaglandin and thromboxane A2 formation (1). N. Engl. J. Med., 1988; 15, 319, 11: 689-98.
69. Ollerenshaw SL, Woolcock AJ: Characteristics of the inflammation in biopsies from large airways of subjects with asthma and subjects with chronic airflow limitation. Am. Rev. Respir. Dis., 1992; 145, 4 Pt 1: 922-7.
70. Openshaw PJ, Hewitt C: Protective and harmful effects of viral infections in childhood on wheezing disorders and asthma. Am. J. Respir. Crit. Care Med., 2000; 162, 2 Pt. 2: 40-43.
71. Paganin F, et al.: Computed tomography of the lungs in asthma: influence of disease severity and etiology. Am. J. Respir. Crit. Care Med., 1996; 153, 1: 110-4.
72. Pattemore PK, et al.: Viruses as precipitants of asthma symptoms. I. Epidemiology. Clin. Exp. Allergy 1992; 22, 3: 325-36.
73. Pawankar R: Epithelial cells as immunoregulators in allergic airway diseases. Curr. Opin. Allergy Clin. Immunol., 2002; 2, 1: 1-5.
74. Pearce N, et al.: Comparison of asthma prevalence in the ISAAC and the ECRHS. ISAAC Steering Committee and the European Community Respiratory Health Survey. International Study of Asthma and Allergies in Childhood. Eur. Respir. J., 2000; 16, 3: 420-6.
75. Peat JK, Li J: Reversing the trend: reducing the prevalence of asthma. J. Allergy Clin. Immunol., 1999; 103, 1 Pt. 1: 1-10.
76. Pirożyński M: Immunopatologia astmy oskrzelowej i przewlekłej obturacyjnej choroby płuc. Pneumonol. Alergol. Pol., 2002, 70, suplement 1: 40-2.
77. Polito AJ, Proud D: Epithelia cells as regulators of airway inflammation. J. Allergy Clin. Immunol., 1998; 102, 5: 714-8.
78. Quanjer PH, et al.: Peak expiratory flow: conclusions and recommendations of a Working Party of the European Respiratory Society. Eur. Respir. J. Suppl., 1997; 24: 2-8.
79. Rhodes HL, et al.: Early life risk factors for adult asthma: a birth cohort study of subjects at risk. J. Allergy Clin. Immunol., 2001; 108, 5: 720-5.
80. Riise GC, et al.: Bronchial brush biopsies for studies of epithelial inflammation in stable asthma and nonobstructive chronic bronchitis. Eur. Respir. J., 1996; 9, 8: 1665-71.
81. Rimpela AH, et al.: Asthma and allergic rhinitis among Finnish adolescents in 1977-1991. Scand. J. Soc. Med., 1995; 23, 1: 60-5.
82. Roche WR, et al.: Subepithelial fibrosis in the bronchi of asthmatics. Lancet 1989; 11, 1, 8637: 520-4.
83. Ronmark E, et al.: Incidence of asthma in adults – report from the Obstructive Lung. Disease in Northern Sweden Study. Allergy 1997; 52, 11: 1071-8.
84. Ryan D: Safety of long-acting beta-agonists. Ann. Intern. Med., 2006; 7, 145, 9: 707-10.
85. Sakula A: Charcot-Leyden crystals and Curschmann spirals in asthmatic sputum. Thorax 1986; 41, 7: 503-7.
86. Sandford A, et al.: The genetics of asthma. Am. J. Respir. Crit. Care Med., 1996; 153, 6 Pt. 1: 1749-65.
87. Sears MR, et al.: The relative risks of sensitivity to grass pollen, house dust mite and cat dander in the development of childhood asthma. Clin. Exp. Allergy 1989; 19, 4: 419-24.
88. Sibbald B, Turner-Warwick M: Factors influencing the prevalence of asthma among first degree relatives of extrinsic and intrinsic asthmatics. Thorax 1979; 34, 3: 332-7.
89. Sporik R, et al.: House dust mite exposure as a cause of asthma. Clin. Exp. Allergy 1992; 22, 10: 897-906.
90. Stolley PD: Asthma mortality. Why the United States was spared an epidemic of deaths due to asthma. Am. Rev. Respir. Dis., 1972; 105, 6: 883-90.
91. Taylor B, et al.: Changes in the reported prevalence of childhood eczema since the 1939-45 war. Lancet 1984; 1, 2, 8414: 1255-7.
92. Toogood JH, et al.: Influence of dosing frequency and schedule on the response of chronic asthmatics to the aerosol steroid, budesonide. J. Allergy Clin. Immunol., 1982; 70, 4: 288-98.
93. Unanue ER, Allen PM: The basis for the immunoregulatory role of macrophages and other accessory cells. Science 1987; 1, 236, 4801: 551-7.
94. Venkayya R, et al.: The Th2 lymphocyte products IL-4 and IL-13 rapidly induce airway hyperresponsiveness through direct effects on resident airway cells. Am. J. Respir. Cell. Mol. Biol., 2002; 26, 2: 202-8.
95. von Mutius E, et al.: Prevalence of asthma and atopy in two areas of West and East Germany. Am. J. Respir. Crit. Care Med., 1994; 149, 2 Pt. 1: 358-64.
96. Warner JA, et al.: Is deficiency of interferon gamma production by allergen triggered cord blood cells a predictor of atopic eczema? Clin. Exp. Allergy 1994; 24, 5: 423-30.
97. Wasserfallen JB, et al.: Sudden asphyxic asthma: a distinct entity? Am. Rev. Respir. Dis., 1990; 142, 1: 108-11.
98. Weiss ST: Diet as a risk factor for asthma. Ciba Found. Symp., 1997, 206: 244-57.
99. Welliver RC: Immunology of respiratory syncytial virus infection: eosinophils, cytokines, chemokines and asthma. Pediatr. Infect. Dis. J.: 2000; 19, 8: 780-3.
100. Wenzel SE, et al.: Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am. J. Respir. Crit. Care Med., 1999; 160, 3: 1001-8.
101. Wenzel SE, et al.: Bronchoscopic evaluation of severe asthma. Persistent inflammation associated with high dose glucocorticoids. Am. J. Respir. Crit. Care Med., 1997; 156, 3 Pt. 1: 737-43.
102. Wiggs BR, et al.: A model of airway narrowing in asthma and in chronic obstructive pulmonary disease. Am. Rev. Respir. Dis., 1992; 145, 6: 1251-8.
103. Wills-Karp M, Chiaramonte M: Interleukin-13 in asthma. Curr. Opin. Pulm. Med., 2003; 9, 1: 21-7.
104. Zhong NS, et al.: Is asymptomatic bronchial hyperresponsiveness an indication of potential asthma? A two-year follow-up of young students with bronchial hyperresponsiveness. Chest 1992; 102, 4: 1104-9.
