© Borgis - Postępy Nauk Medycznych 2/2000, s. 3-7
Piotr Leszczyński1, Jan K. Łącki1, Stefan H. Mackiewicz2
Osteoporoza posteroidowa – patomechanizm, zapobieganie i leczenie
Glucocorticosteroid – induced osteoporosis – patogenesis, prevention and treatment options
1 Klinika Reumatologii i Immunologii Klinicznej Instytutu Chorób Wewnętrznych Akademii Medycznej im. Karola Marcinkowskiego w Poznaniu
Kierownik Kliniki: dr hab. med. Jan K. Łącki
2 Oddział Reumatologii i Centrum Osteoporozy Szpitala im. J. Strusia w Poznaniu
Ordynator Oddziału: prof. Stefan H. Mackiewicz
Summary
Glucocorticosteroid (GSs) has been recognized as well known risk factor for drug induced osteoporosis. Many studies have shown a decrease in bone mass, a bone quality disorders and an increase in the risk of fractures (ribs, forearms and others) in patients with long-term corticosteroid therapy. This is a serious problem concerne large group of patients with connective tissue diseases, rheumatoid arthritis and other systemic diseases. The mechanism of glucocrticoids effects on bone metabolism is complex and leads to increase bone resorption and decrease bone formation. Decrease in bone mass is the most dynamic in the first 6-12 months of exposure to high dose of GSs and require bone loss prevention. Treatment of corticosteroid-induced osteoporosis includs: calcium and vitamin D supplementation, bisphosphonates and as an alternative estrogens, cakitonin or raloxifen.
Słowa kluczowe: osteoporoza,
czynniki ryzyka,
kortykosteroidy,
patomechanizm,
profilaktyka,
leczenie,
postępy nauk medycznych,
wydawnictwo,
czasopismo,
medycyna,
medyczne.

WSTĘP
Glikokortykosteroidy (GKS) od czasów Edwarda C. Kendalla i Filipa S. Hencha, uznanych powszechnie za odkrywców budowy i działania hormonów kory nadnerczy, są powszechnie stosowane we wszystkich działach współczesnej medycyny. Dzięki silnemu działaniu przeciwzapalnemu i immunosupresyjnemu często są lekami z wyboru, mimo licznych działań ubocznych.
Spadek masy kostnej, zaburzenia struktury kości oraz zwiększone ryzyko złamań, szczególnie żeber, kręgów oraz kości przedramienia, są dobrze znanymi powikłaniami osteoporozy posteroidowej i dotyczą 30-50% chorych (16). Wiadomo również, że GKS są lekami najczęściej wywołującymi osteoporozę. Ryzyko jakiegokolwiek złamania w tej grupie chorych wynosi od 8 do 18% i jest 2-3-krotnie większe niż w podobnych grupach osób nie leczonych GKD (14). W Wielkiej Brytanii jest około 250 tys. takich chorych, a tylko 14% jest leczonych zapobiegawczo (9). Podobne dane dotyczące Polski nie są znane.
Powszechnie uważa się, że spadek masy kostnej jest największy i najbardziej dynamiczny w ciągu pierwszych 6-12 miesięcy leczenia średnimi i dużymi dawkami GKS. Wymaga to postępowania profilaktycznego i ewentualnie leczniczego. Następnie proces stabilizuje się i nie jest tak wyraźnie zaznaczony. Ubytek masy kostnej dotyczy przede wszystkim kości beleczkowej (gąbczastej), w mniejszym zaś stopniu kości korowej (zbitej) i wynosi nawet 15% rocznie. Wpływ GKS na rozwój osteopenii i osteoporozy posteroidowej oraz na wzrost ryzyka złamań zależy od dawki dobowej i łącznej, czasu leczenia, sposobu podawania i osobniczej wrażliwości. Prawdopodobnie nie istnieje (brak przekonywujących dowodów) dawka progowa, poniżej której nie dochodzi do rozwoju osteoporozy posteroidowej. Wiele doniesień potwierdza, że dawka GKS w przeliczeniu na prednison poniżej 7,5 mg/dobę jest bezpieczna, szczególnie u kobiet w okresie przedmenopauzalnym i z przewlekłym procesem zapalnym. Natomiast alternatywne podawanie GKS (co drugi dzień) nie ma wpływu na zmniejszenie spadku masy kostnej (4, 10, 11, 14, 16, 18).
PATOGENEZA OSTEOPOROZY POSTEROIDOWEJ
Mechanizm niekorzystnego wpływu GKS na tkankę kostną jest zróżnicowany (ryc. 1) i prowadzi bezpośrednio do zahamowania procesów kościotwórczych, a pośrednio do wzrostu aktywności resorbcyjnej osteoklastów. Zaburzenia homeostazy między osteoblastem a osteoklastem wynikają z wpływu GKS na gospodarkę wapniowo-fosforanową, hormony płciowe oraz wspomniane bezpośrednie hamowanie procesów pobudzających kościotworzenie.
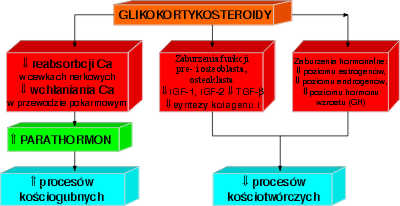
Ryc. 1. Osteoporoza posteroidowa – mechanizm powstawania.
Na poziomie komórkowym GKS hamują replikację i powstawanie komórek linii osteoblastycznej oraz pobudzają ich apoptozę. Utrudniają także ich przyleganie do macierzy kostnej oraz syntezę kolagenu typu I i innych niekolagenowych białek produkowanych przez osteoblasty. Zależnie od dawki stosowanych GKS dochodzi do zmniejszenia aktywności czynników wzrostu (IGF-1, IGF-2, IL-1, TGF-β), zmniejszenie syntezy osteokalcyny, prostaglandyny E oraz zaburzeń ekspresji onkogenów (3, 4, 6, 8).
GKS powodują zmniejszenie wydzielania endogennych hormonów produkowanych przez przysadkę mózgową, przede wszystkim LH (hormon luteinizujący), ale także syntezy androgenów w korze nadnerczy. Bezpośrednio ulega także zahamowaniu produkcja estrogenów w jajnikach i testosteronu w jądrach. Niedobory tych hormonów odgrywają zasadniczą rolę w patogenezie osteoporozy posteroidowej (6, 8, 19, 20 22).
W obrębie przewodu pokarmowego GKS zmniejszają wchłanianie wapnia i fosforanów w jelicie cienkim przez zmniejszenie syntezy CaBP (calcium binding protein – białka wiążącego wapń). Mechanizm ten prawdopodobnie jest niezależny od witaminy D. Jednocześnie w wyniku bezpośredniego działania GKS i ograniczenia reabsorbcji wapnia w cewkach nerkowych dochodzi do hyperkalciurii. Efektem obu procesów jest wtórna nadczynność przytarczyc ze zwiększonym poziomem parathormonu (PTH) w surowicy, co oczywiście nasila proces resorbcji tkanki kostnej (19, 20 22).
Wśród innych mechanizmów prowadzących do utraty masy kostnej podczas długotrwałego leczenia GKS należy podkreślić miopatię posteroidową prowadzącą do osłabienia sił mechanicznych, które fizjologicznie podczas skurczu mięśni obciążają kościec. Oddzielnym zagadnieniem jest osteoporoza wtórna, towarzysząca najczęściej aktywnym chorobom zapalnym (osteoporoza śródzapalna), która nasila wystąpienie spadku masy kostnej u chorych leczonych GKS. Z drugiej strony GKS w tzw. niskich dawkach 5-7,5 mg prednisonu na dobę) skutecznie hamują proces zapalny poprawiając m.in. sprawność chorego, bilans wapniowo-fosforanowy oraz zmniejszają obrót kostny. Co więcej dawki prednisonu poniżej 5 mg/dobę są mało skuteczne w kontroli procesu zapalnego i paradoksalnie odpowiadają za zwiększenie obrotu kostnegi, spadek masy kostnego i przyspieszenie rozwoju osteoporozy (3, 6, 8, 19, 20 22).
BADANIE DENSYTOMETRYCZNE U CHORYCH LECZONYCH GLIKOKORTYKOSTEROIDAMI
Densytometryczne rozpoznanie osteoporozy (ocena gęstości mineralnej kości – BMD) u chorych przewlekle leczonych GKS nie odbiega od ogólnie przyjętych standardów diagnostycznych (7, 12, 17). Najczęściej stosuje się technikę DXA/DEXA (absorbcjometrii promieniowania rentgenowskiego o podwójnej wiązce energetycznej). Miejsce pomiaru w szkielecie powinno uwzględniać znaczną zawartość kości beleczkowej, bardziej aktywnej metabolicznie i wrażliwszej na działanie GKS niż kości korowej.
Najbardziej wiarygodnym miejscem do oceny wpływu kortykoterapii na masę kostną jest odcinek L2-L4 kręgosłupa w projekcji A-P, kość piętowa oraz nROI (obszar w obrębie odcinka dystalnego przedramienia zawierający podobny stosunek kości beleczkowej do korowej jak w części lędźwiowej kręgosłupa). U chorych powyżej 65 roku życia, u których wykonuje się pomiary w obszarze L2-L4 kręgosłupa, należy przy interpretacji badania uwzględnić obecność zmian zwyrodnieniowych, dyskopatii, skrzywienia bocznego z rotacją trzonów kręgowych, a także zmian miażdżycowych w tętnicy brzusznej. Nie zaleca się projekcji bocznej kręgosłupa z powodu braku przewagi tego badania w ocenie ryzyka złamań kręgów.
Wydaje się, że najlepiej wykonać pomiar wyjściowy BMD przed rozpoczęciem każdej kortykoterapii, a na pewno gdy będzie ona trwać powyżej 6 miesięcy. Kontrolne badania densytometryczne należy wykonywać nie częściej niż co 12 miesięcy, chyba że ułatwia to zmianę decyzji terapeutycznych (7, 12, 17, 21).
Pomiarów densytometrycznych ilościową techniką ultradźwiękową (QUS), mimo że prawdopodobnie informują o budowie wewnętrznej kości oraz jej właściwościach materiałowych, nie poleca się jako jedynych w ocenie zmian kostnych. Technikę ultradźwiękową należy traktować jako pomocniczą i o ile to możliwe interpretować łącznie z oceną gęstości mineralnej kości (BMD) (7, 12).
POSTĘPOWANIE PROFILAKTYCZNE I LECZENIE
Optymalna strategia postępowania zapobiegająca utracie masy kostnej spowodowanej kortykoterapią obejmuje stosowanie najniższej skutecznej dawki GKS, preparatów podawanych miejscowo, drogą wziewną lub dożylnie np. metodą pulsacyjną co 4-6 tygodni z przerwą między „pulsami”. Każdego chorego można zachęcać do aktywności fizycznej (ćwiczenia obciążające szkielet przynajmniej przez 30 minut dziennie), zmiany nawyków żywieniowych (dieta bogatowapniowa i ubogobiałkowa, zakaz palenia papierosów oraz ograniczenie picia alkoholu i kawy), a przy braku przeciwwskazań do nasłonecznienia około 15 minut dziennie (22).
U chorych, u których doszło do złamania osteoporotycznego, po przeprowadzeniu szczegółowego wywiadu, badania przedmiotowego i badań pomocniczych umożliwiających rozpoznanie osteoporozy wtórnej, należy przede wszystkim zabezpieczyć chorego lekami przeciwbólowymi. Stosujemy paracetamol w dawkach podzielonych, niesteroidowe leki przeciwzapalne (NLPZ), a także krótko działające opioidowe środki przeciwbólowe. W przypadku dobrej skuteczności przeciwbólowej można stosować kalcytoninę w postaci donosowej lub parenteralnie w dawce 200 j/dobę przez 4-6 tygodni z późniejszym zmniejszeniem dawkowania i ewentualnie odstawieniem leku. Ta grupa chorych powinna znajdować się pod specjalistyczną opieką doskonale wyszkolonych kinezyterapeutów, którzy pokierują ćwiczeniami fizycznymi prowadzącymi do utrzymania lub nawet zwiększenia masy mięśniowej, do zachowania prawidłowej postawy ciała i chodu. W przypadku złamań kręgów należy ograniczyć ruchy pochylania się do przodu oraz dźwiganie, które mogą doprowadzić do dalszych obciążeń i kolejnych złamań kręgosłupa. Należy również rozważyć korzyści i ryzyko dłuższego stosowania gorsetu.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Adachi J.D. et al.: Vitamin D and calcium in the prevention of corticosteroid induced osteoporosis: a 3 year follow up. J. Rheumatol., 1996, 23: 995-1000.
2. Adachi J.D. et. al: Intermittent etidronate therapy to prevent corticosteroid-induced osteoporosis. N. Engl. J. Med., 1997, 337: 382-387.
3. Benvenuti S.: Effect of glucocorticosteroid on bone cell differentiation. Osteoporos Int., 1999, 9: S1.
4. Bianchi G.: Bone densitometry. DEXA. Osteoporos. Int., 9: S4.
5. Buckley L.M. et al.: Calcium and vitamin D3 supplementation prevents bone loss in the spine secondary to low-dose corticosteroids in patients with rheumatoid arthritis: a randomized, dubleblind, placebo-controlled trial. Ann. Inten. Med., 1996, 125: 961:969.
6. Canalis E.: Effects of glucocorticosteroids on bone formation. Osteoporos. Int., 1999, 9: S1.
7. Consensus Development Conference. Who are candidates for preventive and therapeutic therapy of osteoporosis?, Amsterdam 18-23 maja 1996.
8. Dempster D.W.: Effects of glucocorticosteroids on bone resorbtion. Osteoporos. Int., 1999, 9: S1.
9. Dennison E.: Epidemiology of glucocrticoid-induced osteoporosis. Osteoporos. Int., 1999, 9 (suppl1): S16.
10. Felder M., Ruegsegger P.: Bone loss in patients with rheumatoid arthritis – effect of steroids measured by low dose quantitative computed tomography. Rheumatology Int., 1991, 11: 41.
11. Gough A.K. et al.: Generalised bone loss in patients with early rheumatoid arthritis occurs early and relates to disease activity. Lancet, 1994, 344: 23.
12. Gregg E.W. et al.: The epidemiology of quantitative ultrasound: a review of the relationship with bone mass, osteoporosis and fracture risk. Osteoporosis Int., 1997; 7: 89-99.
13. Hall G.M. et al.: Effect of hormone replacement therapy on bone mass in rheumatoid arthritis patients treated with and without steroids. Arthritis Rheum., 1994, 37: 1499-1505.
14. Hahn T.J.: Primer on the metabolic bone disease and disorders of mineral metabolism. Raven Press, New York, 1993: 250.
15. Healey J.H., Paget S.A., Williams-Russo P. et al.: A randomized controlled trial of salomon calcitonin to prevent bone loss in corticosteroid-trated temporal arteritis and polymyalgia rheumatica. Calcif. Tissue Int., 1996, 58: 73-80.
16. Kanis J.A.: Osteoporosis, London, 1997: 92.
17. Kanis J.A. et al.: The Diagnosis of osteoporosis. J. Bone Miner. Res., 1994, Vol. 9 (8): 1137-1141.
18. Leboff M.S. et al.: Low dose prednisone does not effect calcium homeostasis or bone density in postmenopausal women with rheumatoid arthritis. J. Rheumatol., 1991, 18: 339.
19. Leszczyński P. i wsp.: Masa kostna u chorych na reumatoidalne zapalenie stawów. Ocena wpływu niskich dawek glikokortykosteroidów. Post. Osteoartrol., 1998, 10: 46.
20. Leszczyński P. i wsp.: Ocena wpływu przewlekłej glikokortykosteroidoterapii na masę kostną u chorych na reumatoidalne zapalenie stawów. Nowiny Lekarskie, 1997, 66, 2: 189-194.
21. Ravn P. et al.: Bone densitometry: A new highly responsive region of interest in the distal forearm to monitor the effect of osteoporosis treatment. Osteoporos. Int., 1999, 9: 277-283.
22. Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis American College of Rheumatology Task Force on Osteoporosis Guidelines. Arthritis Rheum., 1996, 11: 1791:1801.
23. Roux C., Oriente P., Laan R. et al.: Randomized trial of effect of cyclical etidronate in the prevention of corticosteroid-induced bone loss. J. Clin. Indocrinol. Metab., 1998, 83: 1128-1133.
24. Saag K.G. et al.: Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. N. Engl. J. Med., 1998, 339: 292-299.
25. Sambrook P. et al.: Prevention of corticosteroid osteoporosis: a comparison of calcium, calcitriol, and calcitonin. N. Engl. J. Med., 1993, 328: 1747-1752.
