© Borgis - Postępy Nauk Medycznych 3/2000, s. 40-50
Bożenna Sawicka
Rozsiane wykrzepianie wewnątrznaczyniowe: aspekty kliniczne, laboratoryjne i terapeutyczne
Disseminated intravascular coagulation: clinical, laboratory and therapeutic aspects
Zakład Diagnostyki Laboratoryjnej CMKP
Kierownik Zakładu: prof. dr hab. n. med. Dagna Bobilewicz
Streszczenie
W pracy przedstawiono wiele złożonych patofizjologicznych mechanizmów wykrzepiania wewnątrznaczyniowego (DIC) oraz objawy kliniczne i wyniki badań laboratoryjnych. Najczęściej DIC występuje w różnych rodzajach urazów, komplikacjach położniczych, nowotworach i stanach zapalnych. Wiele decyzji terapeutycznych jest i pozostanie kontrowersyjnymi, aż do opublikowania bardziej dokładnych danych dotyczących przeżycia pacjentów. Aktualnie badane są nowe leki, działające nie tylko przeciwzakrzepowo, ale również wpływające na aktywność cytokin i innych substancji.
Summary
Current concepts of the many complex pathophysiological mechanisms and clinical and laboratory manifestations of disseminated intravascular coagulation (DIC) are presented. Clinical disorders giving rise to DIC fall into categories of trauma, obstetrical complications, malignancies and a variety of inflammatory conditions.
Many therapeutic decisions are controversial and will remain so until more is published about specific therapeutic modalities and survival patterns.
The future holds promise for not only newer antithrombotic agents, but also agents which will block, blunt or modify cytokine activity and the activity of vasoactive substances.

Wprowadzenie
Rozsiane wykrzepianie wewnątrznaczyniowe (DIC) jest nabytym zaburzeniem krzepnięcia krwi pojawiającym się w wyniku zakłócenia równowagi hemostatycznej i nadmiernego tworzenia trombiny.
Trombina jest centralnym enzymem regulacyjnym w hemostazie i wykazuje rozległy zakres biologicznej aktywności (18). Jest ona najsilniejszym, ze znanych, agonistą płytek, tworzy fibrynę, aktywuje różne proenzymy i prokofaktory. Trombina ma również antykoagulacyjne i profibrynolityczne właściwości, może wpływać na uwalnianie prozapalnych cytokin oraz działa jako czynnik wzrostu stymulujący naprawę uszkodzonych tkanek (9, 18). Tworzenie trombiny jest ściśle regulowane przez działanie inhibitorów krzepnięcia zwanych naturalnymi antykoagulantami. Należą do nich: antytrombina III (AT III), której aktywność zwiększa siarczan heparanu, białko C (PC) i jego kofaktor białko S (PS), heparynowy kofaktor II (HC II) oraz inhibitor szlaku czynnika tkankowego (TFPI) (21). Sprawnie działający śródbłonek naczyniowy odsłania na swojej powierzchni receptor trombomodulinę (TM), która wiąże trombinę i osłabia jej działanie prokoagulacyjne. Związanie trombiny w kompleks z trombiną powoduje znaczne nasilenie i przyspieszenie aktywacji białka C. Inne mechanizmy regulujące tworzenie trombiny to: usuwanie z krążenia rozpuszczalnego czynnika tkankowego i rozpuszczalnych kompleksów monomerów fibryny przez komórki jednojądrzaste oraz usuwanie aktywowanych proteaz krzepnięcia i tkankowego aktywatora plazminogenu przez hepatocyty (6). Ważna rola w utrzymaniu prawidłowej hemostazy przypada również układowi fibrynolitycznemu, który przywraca drożność naczyń po utworzeniu złogów fibryny. Ilustrację ww. mechanizmów przedstawiono na rycinie 1.
Układ hemostazy stanowi system złożony, ale i podatny na zaburzenia regulacji. Przy masywnej aktywacji krzepnięcia spowodowanej:
– uszkodzeniem śródbłonka oraz aktywacją płytek, monocytów/makrofagów (posocznice, endotoksemie, infekcje wirusowe),
– wtargnięciem do krwi czynnika tkankowego (operacje, urazy, obumarcie płodu, przedwczesne odklejenie łożyska),
– uszkodzeniem płytek oraz erytrocytów i uwolnieniem z nich czynników trombogennych i zlepiających płytki (przetoczenie krwi niezgodnej grupowo, przełomy hemolityczne, krążenie pozaustrojowe),
– zwolnieniem przepływu krwi w mikrokrążeniu z następczą kwasicą (sinicze wady serca, naczyniaki olbrzymie),
– mechanizmy obronne stają się zbyt słabe i dochodzi do wystąpienia DIC (11). We wcześniejszych doniesieniach zespół DIC określano jako koagulopatię ze zużycia. Było to jednak określenie niezbyt precyzyjne, ponieważ większość składników osocza nie jest zużywana w DIC, lecz są one biodegradowane przez plazminę (5).
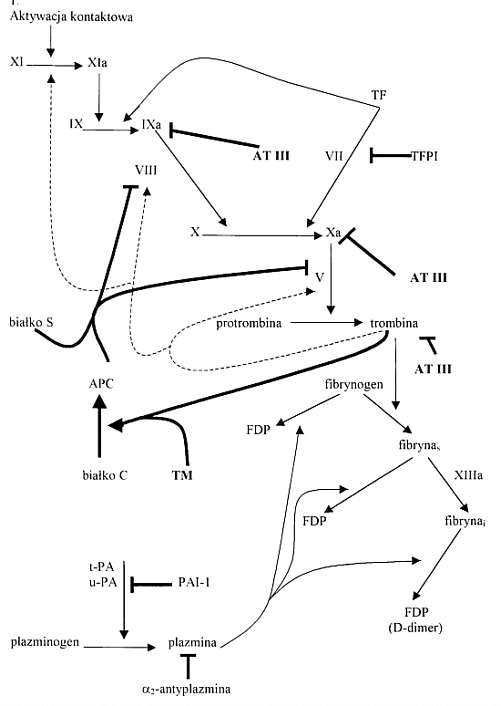
Ryc. 1. Regulacja krzepniecia i fibrynolizy (wg 6). Mechanizmy regulacyjne zostaly uwidocznione przez zastosowanie grubych linii. Linia przerywana przedstawia dodatnie sprzezenie zwrotne (tj. aktywacje czynników V, VIII i XI przez trombine). Znak T oznacza nasilenie reakcji, znak ^ zahamowanie reakcji.
Antykoagulacyjne mechanizmy wplywajace na nasilenie tworzenia trombiny to dzialanie: AT III - antytrombiny III; TFPI - inhibitora szlaku czynnika tkankowego szlaku bialka C (PC) tj.: TM - trombomoduliny i APC - aktywowanego bialka C.
Mechanizmy fibrynolityczne wplywajace na tworzenie plazminy to: t-PA - tkankowy aktywator plazminogenu, u-PA - aktywator plazminogenu typu urokinazowego, PAI-1 - inhibitor aktywatora plazminogenu -1.
Fibryna S - fibryna rozpuszczalna (bez wiazan krzyzowych)
Fibryna i - fibryna nierozpuszczalna (z wiazaniami krzyzowymi).
FDP - produkty degradacji fibryny i fibrynogenu.
Według Bicka (5) terminologia DIC jako zespołu „defibrynacji” powinna raczej być zastąpiona przez określenie zespół „defibrynogenacji”. Bick rozważając nazwę „rozsiane wykrzepianie wewnątrznaczyniowe” uważa, że jest to użyteczne, opisowe, patofizjologiczne określenie – pod warunkiem, że akceptuje ono zarówno stany krwotoczne, jak i zakrzepicę. Ścisła definicja DIC, według ww. autora, to „ogólnoustrojowe zakrzepowo-krwotoczne zaburzenie związane z dobrze określonymi sytuacjami klinicznymi i dokumentacją laboratoryjną:
– aktywacji krzepnięcia Ţ aktywacji fibrynolizy Ţ zużycia inhibitorów i końcowego uszkodzenia narządów lub ich niewydolności.
Etiologia DIC
DIC może towarzyszyć wielu jednostkom i stanom chorobowym (tab. 1). Może mieć przebieg ostry lub przewlekły. Ostre rozsiane wykrzepianie wewnątrznaczyniowe jest zazwyczaj wywołane przez: zakażenia bakteryjne, powikłania położnicze, uraz lub przetoczenie krwinek czerwonych niezgodnych w zakresie ABO (3).
Tabela 1. Przyczyny rozsianego wykrzepiania wewnatrznaczyniowego.
Posocznice
Gram-ujemne
Gram-dodatnie
Wiremie
HIV
Hepatitis
Varicella
Cytomegalia
Powiklania poloznicze
Zator wodami plodowymi
Przedwczesne odklejenie lozyska
Zespól plodu przenoszonego
Zmiazdzenia tkanek
Oparzenia
Hemoliza potransfuzyjna
Rozsiane nowotwory
Bialaczki
Choroby watroby
Ostra niewydolnosc watroby
Ciezka zóltaczka zastoinowa
Operacje serca z zastosowaniem krazenia pozaustrojowego
Protezy naczyniowe
Ukladowe choroby tkanki lacznej
Martwica trzustki
Kwasica cukrzycowa
Sinicze wady serca
Choroby nerek
Naczyniaki olbrzymie
Tetniaki
Amyloidoza
Ukaszenia przez zmije |
Przewlekły DIC – zespół na ogół wyrównanych zaburzeń hemostazy – zwykle związany jest z chorobą nowotworową, obumarłą ciążą, obecnością tętniaków lub naczyniaków (3, 18).
Patofizjologia DIC
W klasycznej koncepcji krzepnięcia krwi trombina tworzona jest przez aktywację szlaku zewnątrzpochodnego (TF – pochodnego) lub wewnątrzpochodnego (zależnego od aktywacji kontaktowej). Ostatnio wykazano, że w większości przypadków DIC inicjacja krzepnięcia zależy od nadmiernego i niekontrolowanego uwalniania czynnika tkankowego (TF). Rola układu wewnątrzpochodnego polega głównie na aktywacji układu kininowego i układu dopełniacza, częściowej aktywacji układu fibrynolitycznego oraz wzmocnieniu krzepnięcia krwi przez niektóre jego czynniki (VIII i IX) (21).
TF może być uwalniany z tkanek uszkodzonych urazem (mechanicznym, cieplnym lub chemicznym), niedokrwieniem, nadmiernym stresem metabolicznym, zakażeniem lub obecnością guzów (2, 3). TF mogą samoczynnie uwalniać komórki białaczkowe oraz nowotworowe (2). Ponadto komórki nowotworowe mogą bezpośrednio aktywować krzepnięcie przez aktywację cz. X, zaś jady węży przez aktywację cz. X i II (układ tenazy i protrombinazy) (11). Krążące leukocyty mogą uwalniać TF w odpowiedzi na działanie endotoksyny, kompleksów immunologicznych lub komórek nowotworowych (ryc. 2). Źródłem TF mogą być również komórki śródbłonka oraz monocyty/makrofagi stymulowane przez takie cytokiny, jak czynnik martwicy nowotworów (TNF) interleukina 1 (IL-1) oraz endotoksynę (3, 9, 12, 21, 24). TF tworzy kompleks z cz. VII i bezpośrednio stymuluje wytwarzanie enzymatycznych składników kompleksu tenazy (czynnik IX) i protrombinazy (czynnik X), które są niezbędne do tworzenia trombiny. Kompleks TFlVIIa zostaje następnie gwałtownie unieczynniony przez inhibitor szlaku czynnika tkankowego (TFPI), ale uaktywniony kompleks tenazy nadal tworzy trombinę. Aktywacja kompleksów tenazy (aktywacja czynnika X przez aktywny czynnik IX przy współudziale kofaktora – czynnika VIII i jonów wapnia) i protrombinazy (aktywacja czynnika II przez aktywny czynnik X przy współudziale kofaktora czynnika V i jonów wapnia) przebiega na powierzchni fosfolipidów (PL). Źródłem PL mogą być aktywowane płytki krwi, monocyty, makrofagi oraz komórki śródbłonka.
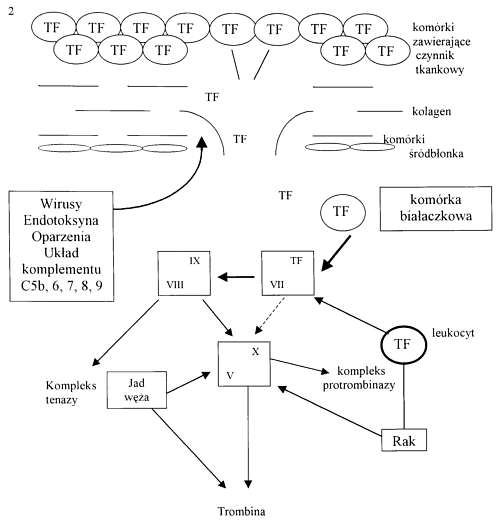
Ryc. 2. Rózne mechanizmy prowadzace do tworzenia trombiny (wg 3). TF - czynnik tkankowy.
Działanie trombiny (bezpośrednie i pośrednie)
Trombina powoduje odcinanie fibrynopeptydów A i B (FA, FB) od fibrynogenu i tworzenie monomerów fibryny. Monomery fibryny polimeryzując ze sobą tworzą fibrynę, co prowadzi do mikro- i makronaczyniowej zakrzepicy, obwodowego niedotlenienia i uszkodzenia narządów (5). W miejscach uszkodzenia śródbłonka i tworzenia złogów fibryny osadzają się płytki krwi, które ulegają agregacji i uwalniają ziarnistości płytkowe. Prowadzi to do małopłytkowości. Nadmiar trombiny może powodować uwalnianie ze śródbłonka trombomoduliny (TM) o dużym znaczeniu diagnostycznym w DIC, endoteliny (ET) i selektyny E (SE) (1, 5, 16, 24). Uwalniana endotelina powoduje intensywne zwężanie naczyń i ich skurcz, co sprzyja zakrzepicy i niedrożności naczyń oraz prowadzi do większego końcowego uszkodzenia narządów (5). Selektyna E wiąże granulocyty, limfocyty i makrofagi do śródbłonka oraz stymuluje uwalnianie cytokin i czynnika aktywującego płytki (PAF) (5).
PAF, który może również pochodzić z aktywowanego przez trombinę śródbłonka, komórek tucznych, eozynofilii i płytek krwi powoduje agregację płytek i nasila trombocytopenię (5). Wiązanie granulocytów do śródbłonka przez SE stymuluje ich aktywację i uwalnianie katepsyny i elastazy. Katepsyna i elastaza, enzymy o niezwykle silnym działaniu, powodują: degradację wielu prokoagulantów, zwiększone uwalnianie cytokin i prowadzą do uszkodzenia narządów (5, 14, 17). Dodatkowo, kallikreina i cz. XIIa mogą nasilać uwalnianie elastazy z granulocytów obojętnochłonnych (14).
Skutki działania plazminy
W zespole wykrzepiania wewnątrznaczyniowego w krążeniu ogólnoustrojowym oprócz trombiny znajduje się plazmina. Powstaje ona z plazminogenu, głównie w wyniku działania tkankowego aktywatora plazminogenu (tPA). Plazmina odcina od fibryny i fibrynogenu fragmenty X, Y, D i E, zwane produktami degradacji (FDP). FDP hamują polimeryzację monomerów fibryny i nasilają skazę krwotoczną. FDP mogą również łączyć się z krążącymi monomerami fibryny i tworzyć rozpuszczalne kompleksy zwane rozpuszczalną fibryną (SF) (ryc. 3). Późne fragmenty degradacji D i E mają wysokie powinowactwo do powierzchni płytek krwi i powodują ważne klinicznie upośledzenie ich funkcji (5). Plazmina, działając na fibrynogen odcina specyficzny peptyd Bb 1-42, zaś działając na fibrynę odcina peptyd Bb 15-42. Oba mają znaczenie diagnostyczne w DIC (5). W wyniku działania plazminy na skrzep fibrynowy stabilizowany przez cz. XIIIa (krzyżowo związana fibryna) powstają specyficzne produkty degradacji zwane D-dimerami (ryc. 4). Plazmina również efektywnie biodegraduje czynniki V, VIIIc, IX, XI, oraz inne osoczowe białka – wśród nich hormon wzrostu, ACTH i insulinę (5). Plazmina może aktywować komponenty C1 i C3 układu dopełniacza z końcową aktywacją C8, 9 i prowadzić do lizy krwinek czerwonych oraz płytek krwi (5). Krwinki czerwone i płytki krwi uwalniając ADP i fosfolipidy z błon komórkowych stają się dodatkowym źródłem materiału prokogulacyjnego. Aktywacja dopełniacza zwiększa przepuszczalność naczyń oraz może prowadzić do hipotensji i szoku (5, 18). Podwyższony poziom inhibitora tkankowego aktywatora plazminogenu (PAI-1) w DIC może tłumić niektóre z tych aktywności, osłabiać aktywność układu fibrynolitycznego i zwiększać tworzenie fibryny (5, 21). W wyniku aktywacji procesu krzepnięcia i fibrynolizy w DIC dochodzi do obniżenia poziomu większości inhibitorów hemostazy (3, 17, 21). Dotyczy to w głównej mierze antytrombiny III, aktywowanego białka C (APC), a2 antyplazminy, w mniejszym stopniu białka S (PS) i heparynowego kofaktora II (HC II). Ważny udział w przebiegu DIC przypada również aktywacji układu kininowego (ryc. 3). Powstała z prekallikreiny kallikreina przekształca wysokocząsteczkowy kininogen do kinin, które zwiększają przepuszczalność naczyń krwionośnych oraz powodują hipotensję i szok (5, 18). Utworzona bradykinina może rozszerzać naczynia krwionośne bezpośrednio lub pośrednio przez wpływ na syntezę tlenku azotu (NO) i prostacykliny (PGI 2) (21).
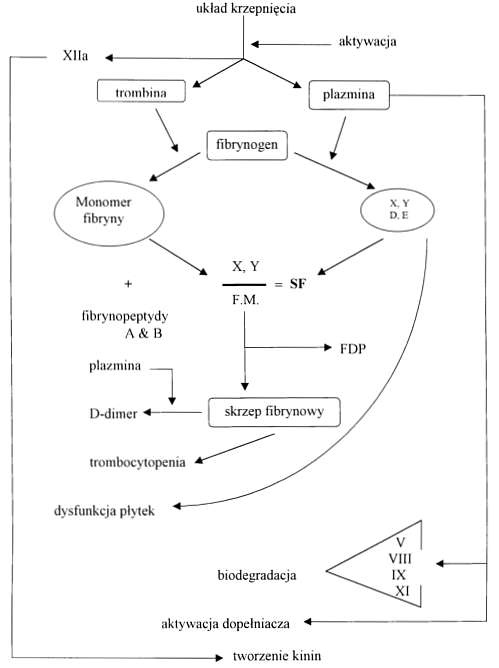
Ryc. 3. Patofizjologia DIC (wg 5). FDP - produkty degradacji fibryny i fibrynogenu; X, Y, D, E - fragmenty X, Y, D, E;
SF - fibryna rozpuszczalna; FM - monomery fibryny.
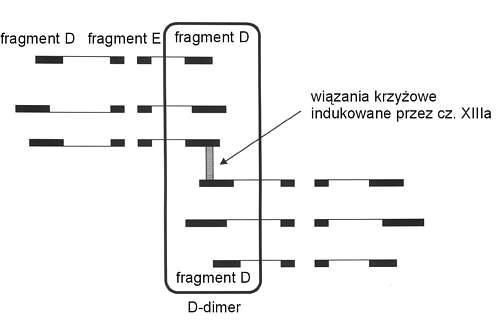
Ryc. 4. Tworzenie D-dimerów (wg 5).
Udział cytokin w DIC
Udział cytokin w mechanizmach patogenetycznych DIC budzi duże zainteresowanie i jest badany intensywnie.
Aktualnie zidentyfikowano ponad 30 cytokin, zaś te, które biorą udział w reakcjach zapalnych ogólnie można podzielić na prozapalne i przeciwzapalne (12). Pojawienie się cytokin prozapalnych w krwioobiegu wykryto w takich stanach patologicznych, jak ciężkie zakażenia, wstrząs septyczny, zespół ostrej niewydolności płuc u dorosłych (ARDS), hemolityczne i niehemolityczne powikłania poprzetoczeniowe, niektóre nowotwory (8, 12, 17, 25). Jak wynika z obserwacji, są to zarazem te stany, w których występuje jawny klinicznie zespół DIC (12).
Wśród cytokin prozapalnych czołową pozycję zajmuje czynnik martwicy nowotworów (TNF) oraz interleukina -1 (IL-1). Cytokiny te, o działaniu prozakrzepowym, indukują syntezę i ekspresję białek adhezyjnych na powierzchni komórek krwi i śródbłonka prowadząc do zatykania mikrokrążenia przez agregaty płytkowe (11, 12). Wykazano również, że TNF i IL-1 stymulują syntezę i ekspresję powierzchniową TF w monocytach, makrofagach i śródbłonku naczyń krwionośnych, co odgrywa ważną rolę w inicjacji krzepnięcia wewnątrznaczyniowego i tworzeniu trombiny (3, 6, 11, 12, 21). Poprzez aktywację kolagenolizy TNF i IL-1 powodują uszkodzenie układu naczyniowego (12). Cytokiny prozapalne mogą drastycznie wpływać na przeciwzakrzepowe właściwości śródbłonka naczyń krwionośnych i powodować uwalnianie trombomoduliny (1, 3, 6, 12, 21, 23). Aktywują one płytki krwi, wywołują ich agregację oraz reakcję uwalniania składników ziarnistości (12). Do cytokin prozapalnych należą również: interleukina 6 (IL-6) oraz interleukina 8 (IL-8). IL-6 stymuluje syntezę białek ostrej fazy, w tym fibrynogenu, oraz trombopoezę (12). IL-8 powoduje przyciąganie leukocytów, ich wywędrowywanie przez ścianę naczyniową i naciekanie tkanek oraz uczula granulocyty na działanie czynników uwalniających zawartość ich ziarnistości (12). Oddziaływania między układami cytokin i hemostazy są obustronne. Prozapalne cytokiny prowokują zmiany w reakcjach hemostatycznych, ale i odwrotnie układ hemostazy wywiera wpływ na tworzenie cytokin (5, 6, 12). Na przykład, powstała w wyniku aktywacji krzepnięcia krwi trombina stymuluje ekspresję TNF, IL-1, IL-6, IL-8 w monocytach, makrofagach i komórkach śródbłonka naczyniowego (5, 12). Fibryna zwiększa ekspresję IL-1 w ludzkich leukocytach, zaś FDP i D-dimery pobudzają syntezę i uwalnianie IL-1 i IL-6 w monocytach (5, 12).
Analizując patofizjologię DIC, staje się zrozumiałe, że objawy kliniczne tego zespołu zależeć będą nie tylko od obecności w krwioobiegu trombiny i plazminy i ich wzajemnych relacji, ale również i od aktywacji innych układów (dopełniacza, kininowego, cytokin).
Rozpoznanie kliniczne
Rozpoznanie zespołu DIC opiera się głównie na obserwacjach klinicznych, a potwierdza się je wykonując odpowiednie badania laboratoryjne. W zależności od szybkości tworzenia i odkładania włóknika w mikrokrążeniu oraz jego degradacji zespół DIC może przebiegać bezobjawowo, mogą występować ciężkie krwawienia lub zakrzepica z objawami niewydolności narządów. Może też występować równocześnie krwawienie i zakrzepica. Nasilone ostre DIC jest najgroźniejszą z nabytych skaz krwotocznych i może mieć bardzo ciężki i gwałtowny przebieg. Najczęstszym i najwcześniej dostrzeganym objawem jest silne krwawienie spowodowane znacznym niedoborem osoczowych czynników krzepnięcia i płytek krwi oraz nadmierną aktywacją fibrynolizy. W ostrym DIC krwawienie przebiega zwykle z objawami hipowolemii, hipotonii i wstrząsu (3). Jest to prawdopodobnie spowodowane aktywacją czynników kontaktu i utworzeniem bradykininy oraz współdziałaniem takich cytokin, jak TNF i IL-1 (3, 12). Obie te cytokiny o działaniu prozapalnym wywołują gorączkę, spadek ciśnienia krwi, przy dużych dawkach zaś – wstrząs, bóle mięśniowe i senność (6, 12, 21).W DIC dochodzi do uszkodzenia i niewydolności wielu ważnych dla życia narządów, których krążenie jest zablokowane mikroskrzeplinami i pojawienia się różnorodnych objawów z tym związanych. Wykrzepianie w naczyniach włosowatych nerek często prowadzi do ostrej martwicy cewek nerkowych lub ostrej niewydolności nerek. Rozsiane wykrzepianie w obrębie naczyń mózgowych powoduje uogólnione zaburzenia czynności kory mózgowej i pnia mózgu z objawami zaburzeń świadomości i śpiączką. Nagłe pogorszenie, któremu towarzyszą zlokalizowane objawy, sugeruje możliwość krwawienia śródczaszkowego. Zakrzepica i krwawienia w obrębie płuc są przyczyną niedotlenienia i ostrej niewydolności oddechowej. Szczególnie ciężką postacią DIC jest piorunująca plamica z krwotoczną martwicą skóry i zgorzelą obserwowane w przebiegu ospy wietrznej lub płonicy u dzieci (3, 21). W przypadkach DIC o przebiegu piorunującym w badaniu sekcyjnym stwierdza się obecność krwotocznej martwicy nadnerczy (zespół Waterhouse?a-Friedrichsena) (3). Schyłkowa niewydolność narządów stanowi główną przyczynę zgonów pacjentów w ostrym wykrzepianiu wewnątrznaczyniowym. Objawy kliniczne pomocne przy rozpoznawaniu DIC przedstawiono w tabeli 2.
Tabela 2. Objawy kliniczne zespolu DIC (wg 3).
Nadmierne wytwarzanie plazminy (krwotok)
- samoistne powstawanie siniaków
- wybroczyny
- krwawienie z przewodu pokarmowego
- krwawienie z ukladu oddechowego
- utrwalone krwawienie z miejsc wklucia
- krwawienie z ran pooperacyjnych
- krwawienie wewnatrzczaszkowe
Nadmierne wytwarzanie trombiny (wykrzepianie)
- niewydolnosc nerek
- spiaczka
- niewydolnosc watroby
- niewydolnosc oddechowa
- martwica skóry
- zgorzel
- zakrzepica zylna
Wytwarzanie cytokin i kinin (wstrzas)
- tachykardia
- obnizenie cisnienia tetniczego
- obrzeki
|
Przewlekłe DIC jest często bezobjawowe i nieprawidłowości mogą być wyłącznie obserwowane w badaniach laboratoryjnych.
Badania laboratoryjne w DIC
W 1988 r. Komitet Badań DIC przy Ministerstwie Zdrowia i Opieki Społecznej w Japonii wyodrębnił kryteria diagnostyczne (system punktowy – score) do rozpoznawania zespołu wykrzepiania wewnątrznaczyniowego (1). Badania laboratoryjne zaproponowane przez ten Komitet to: poziom FDP w surowicy, liczba płytek, stężenie osoczowego fibrynogenu i czas protrombinowy (PT). Nie zawsze jednak wymienione badania są wystarczające do diagnozy DIC. Ich przydatność jest niewielka we wczesnym wykrywaniu DIC, tzw. pre-DIC oraz w przypadkach przewlekłego, skompensowanego DIC.
Ogólnie można stwierdzić, że diagnoza laboratoryjna DIC jest trudna, ponieważ wielu badaniom brak jest odpowiedniej czułości i swoistości. Stosowane badania w rozpoznawaniu DIC, to badania skreeningowe, ogólne, wykrywające molekularne markery aktywacji hemostazy oraz inne, np. służące do oceny poziomu naturalnych antykoagulantów.
Badania skreeningowe i ogólne w DIC
Czas protrombinowy (PT) w DIC jest przedłużony u około 50-75% pacjentów z powodu hipofibrynogemii, obniżenia poziomu cz. V i X oraz obecności FDP (5). Przyczynami prawidłowych lub skróconych czasów PT obserwowanych u około 50% pacjentów z DIC są:
– aktywne czynniki krzepnięcia (trombina i Xa), które mogą przyspieszać tworzenie fibryny,
– wczesne produkty degradacji, które mogą być gwałtownie wykrzepiane przez trombinę (5).
Według Bicka czas PT jest ogólnie niewiarygodny i minimalnie użyteczny w DIC (5). Wydłużony aktywowany czas częściowej tromboplastyny (a PTT) obserwowany u 50-60% pacjentów z DIC wynika z biodegradacji cz. V, VIII c, IX i XI przez plazminę, hipofibrynogenemii oraz obecności FDP. Przyczyny skrócenia lub prawidłowego czasu a PTT (opisywane u 40-50% pacjentów z DIC) są takie same, jak dla czasu PT (5). Czas a PTT jest badaniem mniej wiarygodnym niż PT i minimalnie użytecznym w diagnozowaniu DIC (5). Czas trombinowy (TT) i reptilazowy są często przedłużone w DIC z powodu obecności FDP i hipofibrynogenemii. Mogą one być jednak prawidłowe lub skrócone. Niektórzy autorzy (5) uważają, że ww. badania pośrednio mogą być użyteczne w ocenie aktywacji fibrynolizy. Jeżeli pozostawione skrzepy osocza po wykonaniu badania czasu trombinowego i reptilazowego, ulegną rozpuszczeniu w ciągu 10 minut, świadczy to o znamiennej aktywacji fibrynolizy i obecności plazminy w krążeniu (5). Badania czynników krzepnięcia dostarczają bardzo małej lub żadnej użytecznej informacji dla pacjentów z DIC, głównie z tego powodu, że niedobór jednych czynników może być maskowany przez nadmiar innych (niedobór cz. VIII c przez nadmiar cz. X a) (5). Produkty degradacji fibryny/fibrynogenu (FDP) są podwyższone u 75% pacjentów z DIC (5). Oznaczanie D-dimerów jest bardziej nowoczesną metodą i oceniane jest jako jedno z najbardziej wiarygodnych badań w DIC. Wypada ono nieprawidłowo w 93% przypadków DIC, podobnie jak oznaczanie fragmentów protrombiny F 1+2 (5). Mniejszą wiarygodność mają oznaczania antytrombiny III – dające nieprawidłowe wyniki w 89% oraz fibrynopeptydu A – nieprawidłowe wyniki w 88% przypadków DIC (5). Rozbieżność między wiarygodnością metod oznaczania FDP i D-dimerów może wynikać z kilku przyczyn (5):
– złego wykrzepiania fibrynogenu w próbce surowicy do oznaczania FDP (zawyżenie FDP),
– minimalnej aktywacji wtórnej fibrynolizy, co powoduje głównie uwalnianie fragmentów X, te zaś podobnie jak fragmenty Y, są wykrzepiane przez trombinę i usuwane z badanej surowicy (zaniżenie FDP),
– maksymalnej aktywacji wtórnej fibrynolizy, kiedy nadmiar plazminy powoduje degradację fragmentów D i E (zaniżenie FDP),
– nadmiernego uwalniania granulocytowych proteaz (kolagenazy i elastazy), które również mogą degradować fragmenty D i E i prowadzić do fałszywie ujemnych wyników.
W związku z wymienionymi przyczynami, ujemny wynik oznaczania FDP nie wyklucza diagnozy DIC i dlatego badanie to zostało zastąpione przez oznaczanie D-dimerów.
Poziom fibrynogenu w DIC jest niski u mniej niż połowy chorych (3). Ze względu na fakt, że synteza fibrynogenu jest zwiększona w wielu stanach zapalnych, jego prawidłowy poziom może nie odzwierciedlać zużycia w procesie wykrzepiania wewnątrznaczyniowego (18). Zmiany we krwi obwodowej pacjentów z DIC dotyczą głównie liczby płytek, która jest na ogół obniżona. Nasilenie trombocytopenii może być jednak różne – od bardzo niskich wartości, takich jak 20-30 x 109/L do wyższych niż 100 x 109/L (5). Trombocytopenia rzędu 60 x 109/L często jest już zauważalna przy oglądaniu rozmazów krwi obwodowej pacjentów z DIC (5). Testy badające funkcje płytek takie, jak czas krwawienia i agregacja płytek są zwykle nieprawidłowe z powodu opłaszczenia błony płytek przez FDP oraz częściowego uwalniania z płytek materiału prokoagulacyjnego. Nie należy jednak tych badań wykonywać z powodu małej wartości diagnostycznej i możliwości spowodowania dodatkowego krwawienia (czas krwawienia). Zwiększona przemiana płytek i ich skrócone przeżycie u pacjentów z DIC prowadzą do pojawienia się w krążeniu młodych płytek, które jako duże płytki obserwować można w rozmazach krwi obwodowej (5). W rozmazach krwi obwodowej u 50% pacjentów z DIC o przebiegu gwałtownym widoczne są fragmenty krwinek czerwonych zwane schizocytami (5). Powstają one w wyniku mechanicznego uszkodzenia erytrocytów przez złogi fibryny znajdujące się w mikrokrążeniu. U większości pacjentów z ostrym DIC obserwuje się również łagodną retikulocytozę i łagodną leukocytozę, zwykle skojarzoną z pojawianiem się form niedojrzałych (5).
Markery molekularne i inne rzadziej wykonywane badania w diagnozowaniu DIC
Do najważniejszych markerów aktywacji krzepnięcia (ryc. 5) należą:
– fragment protrombiny F1+2 (F1+2) – marker aktywnego cz. X
– fibrynopeptydy A i B (FA, FB) markery
– kompleksy trombina i antytrombina (TAT) działania
– rozpuszczalna fibryna (SF) trombiny
W zespole DIC z reguły obserwuje się podwyższone poziomy tych markerów.
Najważniejszym markerem aktywacji fibrynolizy (ryc. 6) są kompleksy plazmina – a2 antyplazmina (PAP) oraz D-dimery. Zwiększone poziomy kompleksów PAP i D-dimerów w zespole DIC oraz obniżenie poziomu plazminogenu świadczą o aktywacji układu fibrynolitycznego. D-dimery są markerami aktywacji zarówno krzepnięcia, jak i fibrynolizy. Badanie markerów molekularnych aktywacji hemostazy łącznie z innymi badaniami laboratoryjnymi pozwala na różnicowanie między „dominującą hiperfibrynolizą” a „konsumpcyjną koagulopatią” z ograniczonym tworzeniem plazminy (18).
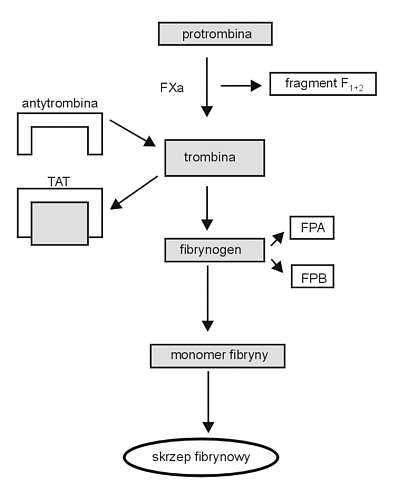
Ryc. 5 Markery aktywacji krzepniecia w DIC. TAT - kompleksy trombina-antytrombina III; FPA - fibrynopeptydy A, FPB - fibrynopeptydy B.
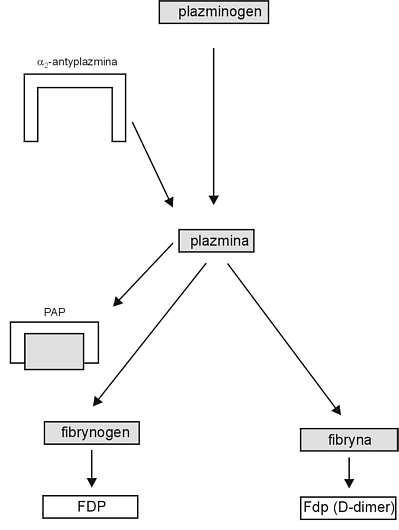
Ryc. 6. Markery aktywacji fibrynolizy w DIC. PAP - kompleksy plazmina-a2 antyplazmina, FDP - produkty degradacji fibryny i fibrynogenu.
Oznaczanie poziomów TAT, PAP, D-dimerów oraz SF ma znaczenie nie tylko w diagnozowaniu zespołu DIC, czy ocenie wyników leczenia, ale również w rozpoznawaniu zaburzeń hemostazy wyprzedzających o kilka dni pojawienie się DIC, tzw. pre-DIC (22, 23, 27, 28). Diagnoza stanu pre-DIC pozwala na wczesne leczenie i zmniejszenie śmiertelności. Niektórzy autorzy przypisują duże znaczenie diagnostyczne oznaczeniu SF, poziom której odzwierciedla aktywne działanie trombiny na fibrynogen (15). SF składa się z kompleksów monomerów i oligomerów fibryny z fibrynogenem i FDP (ryc. 7). Poziom rozpuszczalnej fibryny zależy wyłącznie od tworzenia fibryny, nie zależy zaś od funkcji wątroby, stanów zapalnych czy supresji szpiku kostnego (28). Ma ona dłuższy okres półtrwania w porównaniu z innymi markerami (np. F1+2 lub TAT). Czułość i swoistość SF w diagnozowaniu DIC jest podobna jak dla D-dimerów, ale badanie to jest uważane za bardziej wiarygodne u chorych z osłabioną fibrynolizą. Oznaczanie SF może służyć do oceny skuteczności leczenia antykoagulacyjnego w DIC (6, 28).

Ryc. 7. Powstawanie rozpuszczalnej fibryny (SF), markera nadkrzepliwosci.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Asakura H., Jokaji H., Saito M. i wsp.: Plasma levels of soluble thrombomodulin increase in cases of disseminated intravascular coagulation with organ failure. Am.J.Hematol. 1991, 38, 281-287.
2. Asakura H., Kamikubo Y., Goto A. i wsp.: Role of tissue factor in disseminated intravascular coagulation. Thromb. Res., 1995, 80 (3), 217-224.
3. Baglin T.: Rozsiane wykrzepianie wewnątrznaczyniowe: rozpoznanie i leczenie. BMJ, 1996, 312, 683-7, 43-49.
4. Bajaj M., Bajaj S.P.: Tisue factor pathway inhibitor; potential therapeutic applications. Thromb. Haemost. 1997, 78 (1), 471-477.
5. Bick R.: Disseminated intravascular coagulation: pathophysiological mechanisms and manifestations. Semin. Thromb. Hemost., 1998, 24 (1), 3-18.
6. Carey M., Rodgers G.M.: Disseminated intravascular coagulation: clinical and laboratory aspects. Am. J. Hematol. 1998, 59, 65-73.
7. Gando S., Kameue T., Nanzaki S. i wsp.: Cytokines soluble thrombomodulin and disseminated intravascular coagulation in patients with systemic inflammatory response syndrome. Thromb.Res., 1995, 80 (6), 519-526.
8. Ikegami K., Suzuki Y., Yukoka T. i wsp.: Endothelial cell injury, as quantified by the soluble thrombomodulin level, predicts sepsis/multiple organ dysfunction syndrome after blunt trauma. J.Trauma, 1998, 44 (5), 789-795.
9. Johnson K., Choi Y., De Groot E. i wsp.: Potential mechanisms for a proinflammatory vascular cytokine response to coagulation activation. J.Immunol., 1998, 160, 5130-5135.
10. Katsuura Y., Okamoto S., Ohno N. i wsp.: Effects of a highly selective synthetic inhibitor of plasma kallikrein on disseminated intravascular coagulation in rats. Thromb. Res., 1996, 82 (4), 361-368.
11. Kopeć M.: Skazy krwotoczne w: Choroby wewnętrzne, pod red. F.Kokota, PZWL W-wa, 1996, 576-579.
12. Kopeć M.: Udział cytokin w mechanizmach patogenetycznych rozsianego krzepnięcia śródnaczyniowego. Acta Hematol. Pol., 1997, 28, supl., 59-67.
13. Kunishima S., Kobayashi S., Naol T.: Increased but highy dispersed levels of plasma glycocalicin in patients with disseminated intravascular coagulation. Eur.J.Haematol., 1996, 56, 173-177.
14. Okajima K., Fujise R., Motosato Y. i wsp.: Plasma levels of granulocyte elastase-a1 proteinase inhibitor complex in patients with disseminated intravascular coagulation: pathophysiologic implications. Am.J.Hematol. 1994, 47, 82-88.
15. Okaijma K., Uchiba M., Murakami K. i wsp.: Determination of plasma soluble fibrin using a new Elisa method in patients with disseminated intravascular coagulation. Am.J.Hematol., 1996, 51, 186-191.
16. Okajima K., Uchiba M., Murakami K. i wsp.: Plasma levels of soluble E-selectin in patients with disseminated intravascular coagulation. Am.J.Hematol., 1997, 54, 219-224.
17. Penner J.: Disseminated intravasculare coagulation in patients with multiple organ failure of non-septic origin. Semin.Thromb.Hemost., 1998, 24 (1), 45-52.
18. Riewald M., Riess H.: Treatment options for clinically recognised disseminated intravascular coagulation. Semin.Thromb.Hemost., 1998, 24 (1), 53-59.
19. Shimura M., Wada H., Wakita Y. i wsp.: Plasma tissue factor and tissue factor pathway inhibitor levels in patients with disseminated intravascular coagulation. Am.J.Hematol. 1996, 52 165-170.
20. Takahashi H., Sato N., Shibata A.: Plasma tissue factor pathway inhibitor in disseminated intravascular coagulation comparison of its behavior with plasma tissue factor. Thromb.Res. 1995, 80 (4), 339-348.
21. Vervloet M., Lambertus G., Hack E.: Derangements of coagulation and fibrynolysis in critically ill patients with sepsis and septic shock. Semin.Thromb.Hemost., 1998, 24 (1), 33-34.
22. Wada H., Minamikawa K., Waskita I. i wsp.: Hemostatic study before onset of disseminated intravascular coagulation. Am.J.Hematol., 1993, 43, 190-194.
23. Wada H., Minamikawa K., Wakita Y.: Increased vascular endothelial cell markers in patients with disseminated intravascular coagulation. Am.J.Hematol. 1993, 44, 85-88.
24. Wada H., Mori Y., Shimura M. i wsp.: Poor outcome in disseminated intravascular coagulation or thrombotic thrombocytopenic purpura patients with severe vascular endothelial cell injuries. Am.J.Hematol., 1998, 58, 189-199.
25. Wada H., Ohiwa M., Kaneko T. i wsp.: Plasma level of tumor necrosis factor in disseminated intravascular coagulation. Am.J.Hematol., 1991, 37, 147-151.
26. Wada H. Ohiwa M., Kaneko T. i wsp.: Plasma thrombomodulin as a marker of vascular disorders in thrombotic thrombocytopenic purpura and disseminated intravascular coagulation. Am.J.Hematol., 1992, 39, 20-24.
27. Wada H., Wakita Y., Nakase T. i wsp.: Diagnosis of pre-disseminated intravascular coagulation stage with hemostatic molecular markers. Pol.J.Pharmacol., 1996, 48, 225-228.
28. Wada H., Wakita Y., Nakase T. i wsp.: Increased plasma-soluble fibrin monomer levels in patiensts with disseminated intravascular coagulation. Am.J.Hematol., 1996, 51, 255-260.