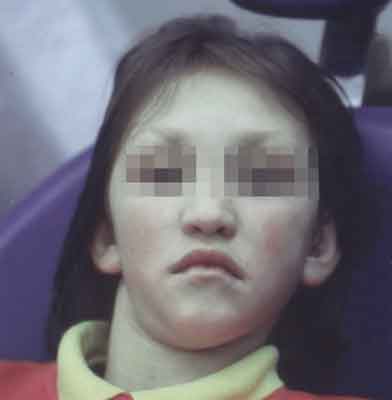
Na podstawie charakterystycznych cech fenotypowych wysunięto rozpoznanie KS. Natomiast występujące u dziewczynki ślady po przetokach na dolnej wardze oraz rozszczep podniebienia były bardziej charakterystyczne dla VDWS.
W 7 roku życia zaobserwowano cechy przedwczesnego dojrzewania płciowego objawiające się powiększeniem gruczołów piersiowych, a pierwsza miesiączka pojawiła się w 9 roku życia (podano leki hamujące przedwczesne dojrzewanie). Badanie ultrasonograficzne jamy brzusznej i echo serca nie wykazały zmian w obrębie narządów wewnętrznych.
Standardowe badanie cytogenetyczne z krwi obwodowej wykazało kariotyp prawidłowy, żeński. Za pomocą techniki FISH wykluczono także mikrodelecję w lokus regionu krytycznego dla VDWS obejmującego gen IRF6 (badanie wykonano w Cen ter for Tuman Genetics, Leuven, Belgia).
W badaniu stomatologicznym zewnątrzustnym, poza powyżej opisanymi cechami twarzoczaszki, stwierdzono wydłużenie dolnego odcinka twarzy przy zachowanej symetrii twarzy, wysunięcie i skrócenie wargi górnej, przy jednoczesnym wygładzeniu bruzdy bródkowo-wargowej. Warga dolna pacjentki była nietypowa: na granicy czerwieni wargi i błony śluzowej obecne były dwa zagłębienia, z których przy ucisku nie wypływała żadna treść (ryc. 2). Oceniając profil pacjentki stwierdzono, że dolny odcinek twarzy jest wydłużony, warga górna wystaje nieco poza przednią granicę pola biometrycznego, a bródka sięga tej granicy (ryc. 3). Analiza ruchów żuchwy wykazała niewielkiego stopnia zbaczanie bródki w lewo podczas odwodzenia żuchwy bez odtwarzania linii pośrodkowej. Ruchy boczne prawidłowe. Staw skroniowo-żuchwowy podczas badania nie wykazywał nieprawidłowości.
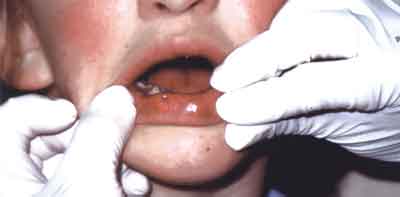
Ryc. 2. Warga dolna pacjentki – widoczne charakterystyczne dla zespołu van der Woude zmiany w postaci zagłębień na granicy czerwieni wargi i błony śluzowej.
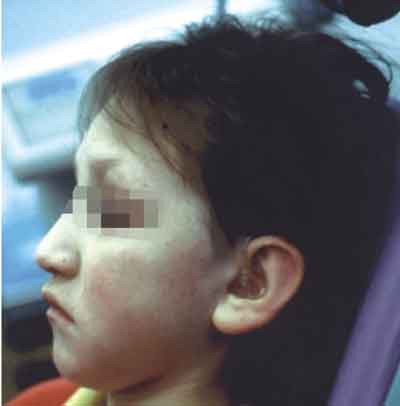
Ryc. 3. Profil pacjentki – widoczne nisko osadzone uszy ze słabo rozwiniętym obrąbkiem, skrócona i wysunięta warga górna, wygładzona bruzda bródkowo-wargowa, kąt między gałęzią i trzonem żuchwy rozwarty.
W badaniu wewnątrzustnym stwierdzono dysfunkcję języka w postaci przetrwałego niemowlęcego typu połykania, podniebienie gotyckie z blizną po operacyjnym zamknięciu rozszczepu po stronie lewej (ryc. 4), zbliżony do paraboli kształt obu łuków zębowych z uzębieniem mieszanym opóźnionym w stosunku do wieku kalendarzowego oraz niekolejne ząbkowanie. Analiza warunków zwarciowych wykazała cechy zgryzu otwartego częściowego przedniego. W obrębie uzębienia zauważono nieprawidłowe ustawienie stałych przyśrodkowych siekaczy w żuchwie, brak stałych bocznych siekaczy w szczęce i żuchwie (w wywiadzie brak trzech mlecznych siekaczy bocznych: 52, 62, 72), nieprawidłowy kształt stałych przyśrodkowych siekaczy w szczęce (stożkowate) z diastemą znacznego stopnia (ryc. 5) oraz zmniejszenie powierzchni żującej pierwszego stałego trzonowca w szczęce. Ubytki próchnicowe stwierdzono w obrębie sześciu zębów (16, 55, 53, 65, 73, 83). Pięć zębów mlecznych (54, 64, 75, 82, 84) zakwalifikowano do ekstrakcji z powodu zgorzelinowego rozpadu miazgi. Dwa pierwsze trzonowce w szczęce były wypełnione. Stan higieny jamy ustnej oceniono jako niedostateczny (OHI = 3).
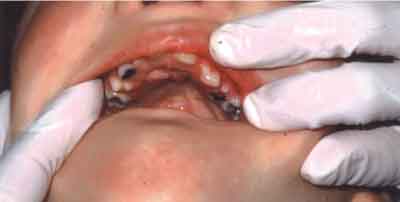
Ryc. 4. Gotyckie podniebienie i blizna po operacji rozszczepu podniebienia.
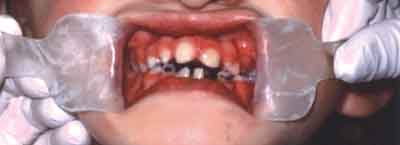
Ryc. 5. Uzębienie pacjentki w zwarciu centralnym: widoczna szpara pionowa, obrócone stałe przyśrodkowe siekacze w szczęce i w żuchwie o nieprawidłowym kształcie, brak stałych siekaczy bocznych w szczęce i w żuchwie.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Niikawa N., et al.: Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J. Pediatr., 1981, 99, 565-569. 2.Kuroki Y., et al.: A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. J. Pediatr., 1981, 99, 570-573. 3.Mc Kusick VA.: Kabuki make-up syndrome. In: Mc Kusick VA, editor. Mendelian inheritance in man. Catalogs of autosomal dominant, autosomal recessive, and X-linked phenotypes (10-th ed.). Baltimore: The Johns Hopkins University Press., 1992 p 633. 4.Niikawa N., et al.: Kabuki make-up (Niikawa-Kuroki) syndrome: a study of 62 patiens. Am. J. Med. Genet., 1988 31, 565-589. 5.Halal F., et al.: Autosomal dominant inheritance of the Kabuki make-up (Niikawa-Kuroki) syndrome. Am. J. Med. Genet., 1989, 33, 376-381. 6.Silengo M., et al.: Sindrome di Niikawa-Kuroki (Kabuki make-up). Descrizione di un caso a probabile ereditarieta autosomica dominante. Riv. Ital. Pediatr., (IJP) 1991, 17, 628-631. 7.Kobayashi O, Sakuragawa N.: Inheritance in Kabuki make-up (Niikawa-Kuroki) syndrome. Am. J. Med. Genet., 1996, 61, 92-93. 8.Tsukahara M., et al.: Dominant inheritance of Kabuki make-up syndrome. Am. J. Med. Genet., 1997, 73, 19-23. 9.Courtens W., et al.: Futher Evidence for Autosomal Dominant Inheritance and Ectodermal Abnormalities in Kabuki Syndrome. Am. J. Genet., 2000, 93, 244-249. 10.Wilson GN.: Thirten cases of Niikawa-Kuroki syndrome: report and review with emphasis on medical complications and preventive management. Am. J. Med. Genet., 1998, 79, 112-120. 11.Lerone M., et al.: Ectodermal abnormalities in Kabuki syndrome. Am. J. Med. Genet., 1997, 73, 263-266. 12.Mhanni AA., Chudley AE.: Genetic landmarks through philately - Kabuki theater and Kabuki syndrome. Clin. Genet., 1999, 56, 116-117. 13. Matsune K., et al.: Caniofacial and dental charakteristics of Kabuki syndrome. Am. J. Med. Genet., 2001, 98, 185-190. 14.Selicorni A., et al.: Biliary atresia and Kabuki syndrome: Another case with long-term follow-up. Am. J. Med. Genet., 2001, 100, 251. 15.Shrandel-Stumpel C., et al.: The Kabuki (Niikawa-Kuroki) syndrome: further delineation of the phenotype in 29 non-Japanisepatients. Eur. J. Pediatr., 1994, 153, 438-445. 16.Chrzanowska K.H., i wsp. Kabuki (Niikawa-Kuroki) syndrome associated with immunodeficiency. Clin. Genet., 1998, 53, 308-312. 17.Wessels MW., et al.: Kabuki syndrome: a review study of three hundred patiens Clin. Dysmorphol., 2002, 11, 95-102. 18.Franceshini, et al.: Lower lip pits and complete idiopathic precocious puberty in a patient with kabuki make-up (Niikawa-Kuroki) syndrome Am. J. Med. Genet., 1993, 47, 423-425. 19.Tsukahara M., et al.: Dominant inheritance of Kabuki make-up syndrome. Am. J. Med. Genet., 1997, 73, 19-23. 20.Chu DC., et al.: CNS malformation in a child with Kabuki (Niikawa-Kuroki) syndrome: report and review. Am. J. Med. Genet., 1997, 72, 205-209. 21.Ewar-Toland A., et al.: Severe congenital anomalies requiring transplantation in children with Kabuki syndrome. Am. J. Med. Genet., 1998, 80, 362-367. 22.Wellesley DG., Stanlej S.: Kabuki make -up and Turner syndromes in the same patient Clin. Dysmorphol., 1994, 3, 727-729. 23.Galan-Gomez E., et al.: Kabuki make-up (Niikawa-Kuroki) syndrome in five Spanish children. Am. J. Med. Genet., 1995, 59, 276-282. 24.Burke LW., Jones MC.: Kabuki syndrome: under-diagnosed recognizable pattern in cleft palate patiens Cleft Palate Craniofac J., 1995, 32, 77-84. 25.Matsune K., et al.: Craniofacial and dental charakteristics of Kabuki syndrome. Am. J. Med. Genet., 2001, 98, 185-190. 26.Mc Kusick VA.: Van der Woude syndrome. In: Mc Kusick VA (wyd) Mendelian inheritance in man. Catalog of human genes and genetic disorders. Twelfth edition. Baltimore: The Johns Hopkins University Press., 1998, s. 381-2 Makita Y., et. al.: Kabuki make-up syndrome is not caused by microdeletion close to the van der Woude syndrome critical region at lq32-q41. Am. J. Med. Genet., 1999, 86, 285-288. 27.Kruk J.: Zespół van der Woude., 1985 Czas. Stom. XXXVII 2, 105-110. 28.Lees M., et al.: Kabuki syndrome with lip pits: a possible microdeletion syndrome involving the van der Woude locus. Am. J. Med. Genet., 1999 36 suppl 1, 5172. 29.Makita Y., et al.: Kabuki make-up syndrome is not caused by microdeletion close to thevan der Woude syndrome critical region at lq32-q41. Am. J. Genet., 1999, 86, 285-288. 30.Kokitsu-Nakata NM., et al.: Lower lip pits anorectal anomalies in Kabuki syndrome. Am. J. Med. Genet., 1999, 86, 282-284. 31.Jackowska M.: Rozpoznawanie - podstawowe jednostki diagnostyczne. Materiały do ćwiczeń z ortodoncji. Szlachetko K. (red.) WAM Warszawa 1993, 69. 32.Zadurska M., i wsp. Hipodoncja - częstość, postacie oraz objawy na podstawie piśmiennictwa i własnego materiału. Czas. Stom., 1999, LII 9 614-621. 33.Remiszewski A.: Przyczynek do zagadnienia dysplazji ektodermalnej u dzieci. Ped. Pol., 1983, 58, 3, 299-305. 34.Mankiewicz M., Bruziewicz-Mikłoszewska B.: Anodoncja, hipodoncja i oligodoncja problemem współczesnej protetyki. Protet. Stom., 1999, 49 5, 254-258. 35.Olczak-Kowalczyk D.: Leczenie protetyczne dzieci w wieku 3-7 lat jako profilaktyka zaburzeń rozwojowych narządu żucia. Protet. Stom., 2002, LII 1 47-54.