© Borgis - Postępy Nauk Medycznych 1/2005, s. 12-17
Andrzej Tykarski, Anna Posadzy-Małaczyńska
Hiperurykemia a nadciśnienie tętnicze
Hyperuricemia and hypertension
z Kliniki Nadciśnienia Tętniczego, Chorób Naczyń i Chorób Wewnętrznych Akademii Medycznej im. Karola Marcinkowskiego w Poznaniu
Kierownik Kliniki: prof. Jerzy Głuszek
Streszczenie
Związek pomiędzy hiperurykemią a nadciśnieniem tętniczym jest znany od dawna. Częstość hiperurykemii w nadciśnieniu tętniczym wynosi według różnych autorów od 3 do 54% i w większości doniesień kilkakrotnie przewyższa analogiczne wartości w populacji ogólnej, które wynoszą od 0,5 do 14%. Większość autorów skłania się ku poglądowi, że hiperurykemia w nadciśnieniu tętniczym jest konsekwencją upośledzenia wydalania nerkowego kwasu moczowego. W nadciśnieniu tętniczym dochodzi do upośledzenia sekrecji kanalikowej kwasu moczowego na skutek zmniejszenia ładunku kwasu moczowego docierającego do przestrzeni okołocewkowej, a stąd do miejsc sekrecji w komórkach cewek nerkowych, na skutek obniżonego nerkowego przepływu krwi. Ponadto stwierdzono, że wzrost poziomu kwasu moczowego i spadek jego klirensu nerkowego wraz z wiekiem jest przyspieszony u osób z nadciśnieniem.
W nadciśnieniu tętniczym leczonym częstość hiperurykemii jest większa (30-58%) w porównaniu z nadciśnienieniem tętniczym nieleczonym (3-38%), co dowodzi wpływu leków hipotensyjnych na poziom kwasu moczowego w surowicy. Leki moczopędne podwyższają poziom kwasu moczowego w surowicy w trakcie przewlekłego leczenia. Leki beta-adrenolityczne powodują również wzrost poziomu kwasu moczowego u chorych na nadciśnienie tętnicze. Efekt ten jest jednak słabszy niż w przypadku stosowania leków moczopędnych i może zanikać w trakcie przewlekłego leczenia. Antagoniści wapnia z nie wpływają na poziom kwasu moczowego w przewlekłym leczeniu hipotensyjnym. Inhibitory konwertazy angiotensyny powodują spadek poziomu kwasu moczowego w surowicy, co wiąże się ze zwiększeniem klirensu nerkowego kwasu moczowego. Silny efekt urikozouryczny oraz redukcję stężenia kwasu moczowego w surowicy stwierdzono również w badaniach klinicznych nad specyficznymi antagonistami receptora angiotensyny AT1, losartanem i valsartanem.
Kwestią nierozstrzygniętą pozostaje, czy hiperurykemia stanowi samodzielny czynnik ryzyka choroby niedokrwiennej serca, niezależny od nadciśnienia tętniczego. Ostatnio wykazano, że w nadciśnieniu tętniczym istnieje zależność pomiędzu indukowanym diuretykiem wzrostem poziomu kwasu moczowego, a ryzykiem wystąpienia choroby niedokrwiennej serca. Raeven zaproponował włączenie hiperurykemii do konstelacji zaburzeń metabolicznych zespołu X.
Summary
Hyperuricemia has long been recognised as a frequent finding in patients with uncomplicated essential hypertension. The prevalence of hyperuricemia in essential hypertension was reported between 5 and 58 % in comparison with 0,5 to 14 % in general population. The most dominant hypothesis attributes the high serum uric acid levels in hypertension to renal vascular and functional impairment. We found a marked decrease in urate excretion per nephron in hyperuricemic patients with essential hypertension and serum uric acid correlated inversely with fractional excretion and tubular secretion of urate. Furthermore it has been shown that serum uric acid elevation and decline in urate clearance with aging is accelerated in hypertensive patients.
It has been suggested that antihypertensive therapy may additionally confound the relationship between hiperuricemia and hypertension. Thiazide diuretic treatment is responsible for increased serum uric in patients with essential hypertension. Other antihypertensive drugs do not affect serum uric acid and angiotensin converting enzyme inhibitors may even prevent development of hyperuricemia in essential hypertension. The increase in serum uric acid during treatment with beta-blockers has not been statistically significant. Nevertheless these drugs decrease tubular secretion of uric acid. Calcium antagonists do not change neither serum uric acid nor its renal excretion. Angiotensin converting enzyme inhibitors indicate uricosuric action. Tubular urate secretion increases in association with improvement of renal hemodynamic parameters. Angiotensin II receptor antagonists are equally effective in decreasing serum uric acid and increasing urate clearance and secretion.
The effect of antihypertensive drugs on serum uric acid was considered of little clinical importance because hyperuricemia was recognized as a marker of early renal vascular involvement in hypertension and a coexisting factor with other independent coronary disease risk factors. Recently positive association between rate of cardiovascular disease and diuretic-induced increases in serum uric acid within the population of tretaed hypertensive patients was reported. It was pointed out that serum uric acid bears an independent and significant association with cardiocascular events and its increase may be in part responsible for lower than expected cardioprotective effect observed in clinical trials of diuretic-based antihypertensive therapy. It was also found tha hyperuricemia is closely linked to a cluster of metabolic and haemodynamic abnormalities called polymetabolic syndrome X.

CZĘSTOŚĆ HIPERURYKEMII W NADCIŚNIENIU TĘTNICZYM
Ponad pół wieku temu Stanton i Freis(1) przedstawili pierwsze spostrzeżenia o podwyższonym poziomie kwasu moczowego w surowicy w nadciśnieniu tętniczym. Związek pomiędzy hiperurykemią a nadciśnieniem tętniczym wzbudził jednak szersze zainteresowanie dopiero w latach 60. po publikacji Duncana (2), który opisał przypadek rodziny, w której u ojca i sześciu z siedmiu synów występowała hiperurykemia, a u matki i wszystkich siedmiu synów nadciśnienie tętnicze. Kolejne doniesienia dotyczące tego problemu, oparte już na dużych grupach chorych, potwierdziły istnienie zależności pomiędzy hiperurykemią a nadciśnieniem tętniczym (3, 4, 5).
Częstość hiperurykemii w nadciśnieniu tętniczym wynosi według różnych autorów od 3 do 54% i w większości doniesień kilkakrotnie przewyższa analogiczne wartości w populacji ogólnej, które wynoszą od 0,5 do 14% (6, 7, 8, 9). W nadciśnieniu tętniczym leczonym częstość hiperurykemii jest większa (30-58%) w porównaniu z nadciśnieniem tętniczym nieleczonym (3-38%), co dowodzi wpływu leków hipotensyjnych na poziom kwasu moczowego w surowicy (3, 4). W nadciśnieniu tętniczym pierwotnym hiperurykemia występuje z podobną częstością (37-43%) jak w nadciśnieniu naczyniowo-nerkowym (44-45%), co przemawia z kolei przeciwko patogenetycznej roli kwasu moczowego w rozwoju nadciśnienia tętniczego (5, 10). Cannon i wsp. (5) stwierdzili hiperurykemię u 75% chorych z nadciśnieniem tętniczym złośliwym. Inni autorzy wykazali korelację pomiędzy stężeniem kwasu moczowego, a ciśnieniem tętniczym skurczowym i rozkurczowym.
ZALEŻNOŚĆ PATOGENETYCZNA: HIPERURYKEMIA – NADCIŚNIENIE TĘTNICZE
Większość autorów skłania się ku poglądowi, że hiperurykemia w nadciśnieniu tętniczym jest konsekwencją upośledzenia wydalania nerkowego kwasu moczowego (3, 5, 10). Zmiany hemodynamiczne w nerkach (11), zwiększone stężenie kwasu mlekowego (12), zmniejszenie przesączania kłębuszkowego (4), nasilenie przeciwtransportu sodowo-litowego (13), wzrost reabsorpcji zwrotnej sodu (14), insulinooporność (15) i leczenie hipotensyjne (16) to proponowane czynniki odpowiedzialne za spadek klirensu kwasu moczowego w nadciśnieniu tętniczym.
Najbardziej prawdopodobna jest hipoteza, która zakłada, że u podstaw hiperurykemii w nadciśnieniu tętniczym leżą zaburzenia hemodynamiczne funkcji nerek (17). Zgodnie z tą teorią w nadciśnieniu tętniczym dochodzi do wzrostu oporu obwodowego, a szczególnie oporu wewnątrznerkowego, który prowadzi do obniżenia nerkowego przepływu krwi, zwłaszcza przepływu korowego, wywołując zaburzenia w transporcie cewkowym kwasu moczowego. W konsekwencji klirens nerkowy kwasu moczowego ulega obniżeniu, a jego stężenie we krwi wzrasta. W badaniach chorych z jednostronnym zwężeniem tętnicy nerkowej stwierdzono znaczny spadek klirensu kwasu moczowego po stronie niedokrwionej. Chirurgiczna rewaskularyzacja prowadziła do całkowitej normalizacji klirensu (10). Messerli i wsp.(18) wykazali u pacjentów z nadciśnieniem tętniczym granicznym oraz nadciśnieniem tętniczym utrwalonym ujemną korelację pomiędzy poziomem kwasu moczowego w surowicy a nerkowym przepływem krwi. Wspomniani autorzy stwierdzili istotnie wyższy opór wewnątrznerkowy u chorych z nadciśnieniem tętniczym i towarzyszącą hiperurykemią niż u pacjentów z prawidłowym poziomem kwasu moczowego.
Badania ostatnich lat pozwoliły wyjaśnić charakter zaburzeń w transporcie cewkowym kwasu moczowego występujących w nadciśnieniu tętniczym. Transport kwasu moczowego w nefronie jest zjawiskiem złożonym. Powszechnie akceptowany jest 4-fazowy model Sorensena i Levinsona (19) przedstawiony na rycinie 1. Zgodnie z nim kwas moczowy ulega całkowitej filtracji kłębuszkowej. Ładunek filtracyjny jest następnie niemal całkowicie wchłaniany zwrotnie. Etap ten nazywamy reabsorpcją przedsekrecyjną. W dalszej części nefronu kwas moczowy podlega sekrecji cewkowej w ilości wynoszącej ok. 40-50% ładunku filtrowanego. Wreszcie dystalnie do miejsca sekrecji zachodzi reabsorpcja posekrecyjna około 75-80% wydzielonej przez cewki ilości kwasu moczowego.
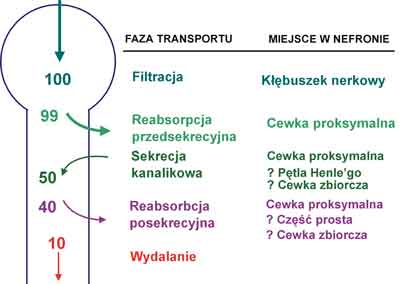
Ryc. 1. Transport kwasu moczowego w nefronie.
W nadciśnieniu tętniczym dochodzi właśnie do upośledzenia sekrecji kanalikowej kwasu moczowego z zachowaniem prawidłowej jego reabsorpcji przedsekrecyjnej i posekrecyjnej (20). Przyczyną obniżonej sekrecji kanalikowej kwasu moczowego w nadciśnieniu tętniczym jest zmniejszenie ładunku kwasu moczowego docierającego do przestrzeni okołocewkowej, a stąd do miejsc sekrecji w komórkach cewek nerkowych, na skutek obniżonego nerkowego przepływu krwi. W nadciśnieniu tętniczym stwierdzono dodatnią korelację pomiędzy klirensem i sekrecją kanalikową kwasu moczowego a nerkowym przepływem krwi (17).
Jednocześnie badania ostatnich lat wykazały zależność wydalania kwasu moczowego od natężenia przeciwtransportu sodowo-litowego w krwinkach czerwonych (13), reabsorpcji zwrotnej sodu (14), stopnia insulinooporności (15) i procesu starzenia, a więc czynników odgrywających rolę patogenetyczną w różnych postaciach nadciśnienia tętniczego. Najbardziej prawdopodobne mechanizmy rozwoju hiperurykemii w nadciśnieniu tętniczym przedstawia rycina 2.
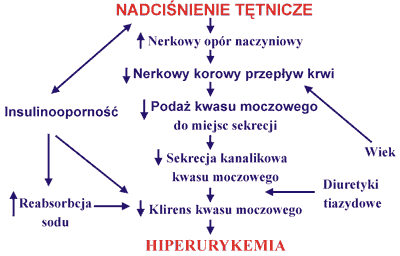
Ryc. 2. Mechanizm hiperurykemii w nadciśnieniu tętniczym.
Inne koncepcje związku hiperurykemii z nadciśnieniem tętniczym
– Rozwój nefropatii moczanowej w przebiegu hiperurykemii prowadzi do nadciśnienia Tętniczego (21)
– Zaburzenia czynności rdzenia nerek na skutek obecności złogów kwasu moczowego z odczynem zapalnym w tkance śródmiąższowej prowadzą do nadciśnienia tętniczego (5)
– Nadużywanie alkoholu jest wspólną przyczyną hiperurykemii i nadciśnienia tętniczego (22)
– W nadciśnieniu tętniczym ciężkim, z powikłaniami narządowymi, stwierdzono, obok redukcji wydalania, nasiloną produkcję kwasu moczowego, prawdopodobnie na skutek upośledzenia perfuzji tkankowej i niedotlenienia (23).
ZNACZENIE KLINICZNE HIPERURYKEMII W NADCIŚNIENIU TĘTNICZYM I INNYCH CHOROBACH UKŁADU KRĄŻENIA
Podwyższone stężenie kwasu moczowego może odgrywać rolę w rozwoju i przebiegu klinicznym nadciśnienia tętniczego. Selby i wsp.(24) w badaniach prospektywnych grupy 2062 hipertoników i normotoników wykazali dodatnią korelację pomiędzy stężeniem kwasu moczowego w surowicy a rozwojem nadciśnienia tętniczego, która była niezależna od innych czynników (waga ciała, dystrybucja tkanki tłuszczowej, poziom kreatyniny, spożycie alkoholu i sodu oraz wywiad rodzinny w kierunku nadciśnienia). Również badania Stamlera (25) nad populacją Chicago wykazały, że stężenie kwasu moczowego w surowicy jest niezależnym czynnikiem wpływającym na późniejszy rozwój nadciśnienia tętniczego.
Badania prospektywne wykazały, że podwyższony poziom kwasu moczowego w surowicy kojarzy się z większą częstością powikłań sercowo-naczyniowych w nadciśnieniu tętniczym (26). Istnieją dwa możliwe wytłumaczenia tej obserwacji: albo hiperurykemia stanowi samodzielny czynnik zagrożenia chorobą wieńcową, albo towarzyszy przypadkom nadciśnienia tętniczego z zaawansowanymi zmianami naczyniowymi. Stwierdzono dodatnią zależność pomiędzy poziomem kwasu moczowego a nerkowym i obwodowym oporem naczyniowym oraz stopniem zaawansowania zmian na dnie oka u chorych z nadciśnieniem tętniczym (20). Chorzy na nadciśnienie tętnicze z towarzyszącą hiperurykemią wykazują w większym stopniu przerost lewej komory w porównaniu z hipertonikami z normourykemią (27). Wydaje się, że poziom kwasu moczowego w surowicy stanowi wczesny marker tendencji do szybkiego rozwoju zmian sercowo-naczyniowych w przebiegu nadciśnienia tętniczego.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Stanton J.R., Freis E.D.: Serum uric acid concentration in essential hypertension. Proc. Soc. Exp. Biol. Med. 1947, 66, 193-200.
2. Duncan H.: Gout, familiar hyperuricemia and renal disease. Quart. J. Med. 1960, 113, 127-129.
3. Breckenridge A.: Hypertension and hyperuricemia. Lancet, 1966, 1, 15-18.
4. Bulpitt C.J.: Serum uric acid in hypertensive patients. Br. Heart J. 1975, 37, 1210-1215.
5. Cannon P.J. et al.: Hyperuricemia in primary and renal hypertension. N. Engl. J. Med. 1966, 275, 457-462.
6. Grajek S. i wsp.: Nadciśnienie tętnicze u mężczyzn w wieku 40-59 lat. Cz.III. Średnie stężenie kwasu moczowego w surowicy hiper- i normotoników. Zależność kwasu moczowego od ciśnienia tętniczego, czasu trwania choroby, wieku, cholesterolu i ciężaru ciała. Kard. Pol. 1983, 26, 149-154.
7. Schrade W. et al.: Humoral changes in atherosclerosis: investigation on lipids, fatty acids, ketones bodies, pyruvic acid and glucose in blood. Lancet, 1960, 2, 409-412.
8. Grayzel A.I. et al.: Diagnostic significance of hyperuricemia in arthritis. N. Engl. J. Med. 1961, 265, 763-769.
9. Itskowitz H.D., Sellers A.: Gout and hyperurycemia after adrenalectomy for hypertension. N. Engl. J. Med. 1963, 268, 1105-110.
10. Simon N.M. et al.: Differential uric acid excretion in essential and renal hypertension. Circulation, 1969, 39, 121-128.
11. Editorial: Hypertension and uric acid. Lancet, 1981, 1, 365-368.
12. Demartini F.E., Cannon P.J., Stason W.B., Laragh J.H.: Lactic acid metabolism in hypertensive patients. Science, 1965, 148, 1482-1488.
13. Strazzullo P., et al.: Red blood cell sodium-lithium countertransport, blood pressure and uric acid metabolism in untreated healthy men. Am. J. Hypertension, 1989, 2, 634-638.
14. Cappuccio P., et al.: Uric acid metabolism and tubular sodium handling. JAMA, 1993, 3, 270-276.
15. Facchini F., et al.: Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. JAMA, 1991, 266, 3008-3012.
16. Barsotti G., et al.: Serum uric acid in mild essential hypertension. Clin. Nephrol. 1983, 45, 145-149.
17. Tykarski A.: Evaluation of renal handling of uric acid in essential hypertension: hyperuricaemia related to decreased urate secretion. Nephron, 1991, 59, 364-368.
18. Messerli F.H., et al.: Serum uric acid in essential hypertension: An indicator of renal vascular involvement. Ann. Intern. Med. 1980, 23, 817-821.
19. Levinson D.J., Sorensen L.B.: Renal handling of uric acid in normal and gouty subjects: evidence for a 4-component system. Ann. Rheum. Dis. 1980, 39, 173-179.
20. Tykarski A.: Mechanizm hiperurykemii oraz wpływ leków hipotensyjnych na transport kwasu moczowego i jego prekursorów w nefronie w nadciśnieniu tętniczym pierwotnym. Praca habilitacyjna. Wydawnictwo Uczelniane Akademii Medycznej w Poznaniu. Poznań 1997.
21. Verger D.: Les tophus gotteux de la medullaire renale des uremiques. Nephron, 1967, 4, 356-360.
22. Ramsey L.E.: Hyperuricemia in hypertension: role of alcohol. Br. Med. J. 1979, 1, 653-659.
23. Łopatka P.: Mechanizm hiperurykemii w nadciśnieniu tętniczym ciężkim i opornym na leczenie. Praca doktorska. Akademia Medyczna w Poznaniu, Poznań 2000.
24. Selby J.V., et al.: Precursors of essential hypertension: Pulmonary function, heart rate, uric acid, serum cholesterol, and other serum chemistries. Am. J. Epidemiol. 1990, 131, 1017-1027.
25. Stamler J.: Relationship of multiple variables of blood pressure. Epidemiology and Control of Hypertension. Georg Thieme Publishers, Stuttgart, 1975, 307-311.
26. Tofuku Y., et al.: Hyperuricemia associated with hypertension. A 4 year follow-up study of hyperuricemic hypertensives. Jap. Circul. 1978, 42, 871-878.
27. Tykarski A.: Zależność pomiędzy stężeniem kwasu moczowego w surowicy a zaawansowaniem zmian naczyniowych i przerostu mięśnia sercowego w nadciśnieniu tętniczym pierwotnym. Pol. Arch. Med. Wewn. 1991, 86, 183-189.
28. Culleton B.F., et al.: Serum uric acid and risk for cardiovascular disease and dath: The Framingham Heart Study.Ann Intern Med. 1999;131:7-13
29. Ostrander L.D., Lamphiear D.E.: Coronary risk factors in a community. Circulation, 1976, 53, 152-156.
30. Coronary Drug Project Research Group: Serum uric acid: Its association with other risk factors and with mortality in coronary heart disease. J. Chronic Dis. 1976, 29, 557-561.
31. Klein R., et al.: Serum uric acid. Its relationship to coronary heart disease risk factors and cardiovascular disease. Arch. Intern. Med. 1973, 132, 401-409.
32. Hall A.P.: Correlation among hyperuricaemia, hypercholesterolemia, coronary disease and hypertension. Arth. Rheum. 1964, 8, 846-852.
33. Benedek T.G.: Correlation of serum uric acid and lipid concentration in normal, gouty and atherosclerotic men. Ann. Intern. Med., 1967, 66, 85-91.
34. Scott J., Sturge R.A.: The effects of weight loss on plasma and urinary uric acid and lipid levels. Adv. Exp. Med. Biol. 1977, 76B, 274-277.
35. Mustard J.F., et al.: Blood coagulation and platelet economy in subjects with primary gout. Can. Med. Assoc. J. 1963, 89, 1207-1212.
36. Ginsberg M.H., et al.: Release of platelet constituents by monosodium urate crystals. J. Clin. Invest. 1977, 60, 999-1005.
37. Godin M.: Hypertension and hyperuricemia: quelle relations? Nouv. Presse Med. 1982, II, 643-649.
38. Fridovich I.: Quantitative aspects of the production of superoxide anion radical by milk xanthine oxidase. J. Biol. Chem. 1970, 245, 4053-4059.
39. Bickel C., et al.: Serum uric acid as an independent predictor of mortalty in patients with angiographically proven coronary artery disease. Am. J. Cardiol. 2002, 89, 12-17.
40. Becker B.F., Reinholz N.: Uric acid as radical scavenger and antioxidant in the heart. Pfligers Arch. 1989, 415, 127-132.
41. Wong K.Y.K., et al.: Urate preicts subsequent cardiac death in stroke survivors. Eur. Heart J. 2002, 23, 788-793.
42. Anker SD., et al.: Uric acid and survival in chronic heart failure. Validation and application in metabolic, functional, and hemodynamic staging. Circulation 2003, 107:1991-1997.
43. Hare J.M, Johnson R.J.: Uric Acid predicts clinical outcomes in heart failure insights regarding the role of xanthine oxidase and uric acid in disease pathophysiology. Editorial. Circulation 2003, 107:1951-1953.
44. Bengtsson C.: Elevated serum uric acid levels during treatment with antihypertensive drugs. Acta Med. Scand. Suppl. 1979, 628, 69-71.
45. Tykarski A. et al.: Mechanism of hyperuricaemic action of angiotensin converting enzyme inhibitors in patients with essential hypertension. J. Hypertension, 1996, 14, Suppl. 1, S219.
