© Borgis - Postępy Nauk Medycznych 1/2009, s. 4-10
*Anna Posadzy-Małaczyńska, Andrzej Tykarski
Hiperurykemia a nadciśnienie tętnicze
Hyperuricemia and hypertension
Klinika i Katedra Nadciśnienia Tętniczego, Chorób Naczyń i Chorób Wewnętrznych Uniwersytetu Medycznego im. Karola Marcinkowskiego w Poznaniu
Kierownik Kliniki: prof. dr hab. med. Jerzy Głuszek
Streszczenie
Częstość hiperurykemii w nadciśnieniu tętniczym wynosi według różnych autorów od 3 do 54% i w większości doniesień kilkakrotnie przewyższa analogiczne wartości w populacji ogólnej, które wynoszą od 0,5 do 14%. Najbardziej prawdopodobna jest hipoteza, która zakłada, że u podstaw hiperurykemii w nadciśnieniu tętniczym leżą zaburzenia hemodynamiczne funkcji nerek, które prowadzą do obniżenia nerkowego przepływu krwi, zwłaszcza przepływu korowego, wywołując zaburzenia w transporcie cewkowym kwasu moczowego. Zmniejszenie perfuzji nerek u chorych z nadciśnieniem tętniczym nakłada się na zmiany postępujące z wiekiem i prowadzi do nasilenia produkcji kwasu moczowego. Wydaje się, że stężenie kwasu moczowego w surowicy stanowi wczesny marker tendencji do szybkiego rozwoju zmian sercowo-naczyniowych w przebiegu nadciśnienia tętniczego. Podwyższenie stężenia kwasu moczowego w surowicy przyczynia się do tworzenia zmian miażdżycowych w naczyniach nie tylko poprzez zwiększenie agregacji płytek czy wpływ mitogenny na komórki śródbłonka, lecz także przez modyfikację jego funkcji. Dane z badań obserwacyjnych i epidemiologicznych wskazują na hiperurykemię jako niezależny czynnik ryzyka nadciśnienia tętniczego. Pomimo patofizjologicznych przesłanek obserwacje rodzą pytanie: czy można zapobiec lub oddalić wystąpienie nadciśnienia i jego powikłań naczyniowych poprzez redukcję stężenia kwasu moczowego w surowicy krwi?
Summary
Hyperuricemia has been found to predict the development of hypertension in numerous studies and to be present more often in adults with hypertension (3–54%) incomparison with normotensive population where are the values of 0.5 do 14%. Some authors have suggested that hyperuricemia may be caused by the reduced renal blood flow as a characteristic hemodynamic finding in hypertension. There is strong evidence that renal vasoconstriction results in increased proximal urate reabsorption and an increase in serum uric acid. The positive association between serum uric acid and hypertension was observed earlier, but in the past it has been unclear whether hyperuricemia played a causal role in arterial hypertension or if it was merely a marker of an underlying pathophysiological process. In several studies serum uric acid has been positively associated with incident hypertension. A number of findings suggest that serum uric acid level can be a strong predictor of cardiovascular disease when combined with elevated blood pressure. Several pathophysiological mechanisms linking serum uric acid to cardiovascular damage at the cellular and tissue level have been proposed. In addition, uric acid has proved to be an excellent marker for tissue ischemia and endothelial dysfunction, and it has been shown to play a role in the development of atherosclerotic lesions.
More research need to be done concerning the physiological and clinical consequences of hyperuricemia, and potential effect of lowering uric acid on the development of hypertension, particularly on prevention of the development of cardiovascular injuries.

Wstęp
Kwas moczowy jest u ludzi najważniejszym końcowym produktem przemiany zasad purynowych. Jego bezpośrednimi prekursorami są oksypuryny: hipoksantyna i ksantyna, utleniane wskutek działania oksydazy ksantynowej. Innymi ważnymi zasadami purynowymi są adenina i guanina. Zasady te w wyniku połączenia z pentozą i fosforylacją dają początek nukleotydom stanowiącym istotną rolę w procesach metabolicznych człowieka. Są one niezbędne do przechowywania i przekazywania informacji genetycznej (RNA, DNA), transmisji sygnalizacji komórkowej (GTP, cAMP, cGMP), gromadzenia energii (ATP) i budowy koenzymów (NAD, NADP). Mechanizm reutylizacji zasad purynowych odgrywa ważną rolę, ponieważ zapobiega stratom energetycznym i zapewnia stałą odnowę nukleotydów. Zasady purynowe, które nie weszły do procesu reutylizacji ulegają dalszym przemianom prowadzącym do powstania kwasu moczowego. Inozyna jest katabolizowana do hipoksantyny, a guanina do ksantyny. Końcowym etapem jest reakcja katalizowana przez oksydazę ksantynową hipoksantyny do ksantyny, a tej do kwasu moczowego (1). W tej sytuacji kwas moczowy może być traktowany jako wskaźnik „kryzysu energetycznego” komórki (2), a w warunkach przewlekłej hipoksji dochodzi do znamiennego jego wzrostu (3).
Częstość hiperurykemii w nadciśnieniu tętniczym
Ponad pół wieku temu Stanton i Freis (4) przedstawili pierwsze spostrzeżenia o podwyższonym poziomie kwasu moczowego w surowicy w nadciśnieniu tętniczym. Związek pomiędzy hiperurykemią a nadciśnieniem tętniczym wzbudził jednak szersze zainteresowanie dopiero w latach 60-tych, po publikacji Duncana (5), który opisał przypadek rodziny, w której u ojca i 6. z 7. synów występowała hiperurykemia, a u matki i wszystkich 7. synów nadciśnienie tętnicze. Kolejne doniesienia dotyczące tego problemu, oparte już na dużych grupach chorych potwierdziły istnienie zależności pomiędzy hiperurykemią a nadciśnieniem tętniczym (6, 7, 8).
Częstość hiperurykemii w nadciśnieniu tętniczym wynosi według różnych autorów od 3 do 54% i w większości doniesień kilkakrotnie przewyższa analogiczne wartości w populacji ogólnej, które wynoszą od 0,5 do 14% (9, 10, 11, 12). W nadciśnieniu tętniczym leczonym częstość hiperurykemii jest większa (30-58%) w porównaniu z nadciśnienieniem tętniczym nieleczonym (3-38%), co dowodzi wpływu leków hipotensyjnych na poziom kwasu moczowego w surowicy (6, 7). W pierwotnym nadciśnieniu tętniczym hiperurykemia występuje z podobną częstością (37-43%) jak w nadciśnieniu naczyniowo-nerkowym (44-45%), co przemawia z kolei przeciwko patogenetycznej roli kwasu moczowego w rozwoju nadciśnienia tętniczego (5, 10).
Cannon i wsp. (8) stwierdzili hiperurykemię u 75% chorych z nadciśnieniem tętniczym złośliwym. Inni autorzy wykazali korelację pomiędzy stężeniem kwasu moczowego, a ciśnieniem tętniczym skurczowym i rozkurczowym, a także hipotensyjny wpływ leku obniżającego w surowicy stężenie kwasu moczowego (inhibitora oksydazy ksantyny-allopurinolu) (13).
Potwierdzeniem związku kwasu moczowego z nadciśnieniem tętniczym są także obserwacje dotyczące dzieci z niską masą urodzeniową, które wraz z wiekiem cechowały się podwyższeniem stężenia kwasu moczowego w surowicy, upośledzeniem funkcji śródbłonka i wzrostem ciśnienia tętniczego (14). Inni autorzy zaobserwowali, że hyperurykemia zwiększa ryzyko rozwoju nadciśnienia i jego powikłań u osób normotensyjnych (15, 16) i może być przesłanką do rozwoju nadciśnienia tętniczego.
Przesłanki patofizjologiczne: hiperurykemia – nadciśnienie tętnicze
Niektórzy autorzy na podstawie badań doświadczalnych, zaproponowali model indukowania nadciśnienia poprzez hiperurykemię (17). W literaturze jednak pojawiło się pytanie, czy kwas moczowy jest niezależnym czynnikiem ryzyka rozwoju miażdżycy czy też wpływa na nią jako jeden z elementów zespołu metabolicznego i tą drogą bierze udział w jej tworzeniu (18, 19, 20).
Większość autorów skłania się ku poglądowi, że hiperurykemia w nadciśnieniu tętniczym jest konsekwencją upośledzenia wydalania nerkowego kwasu moczowego (6, 8, 21). Zmiany hemodynamiczne w nerkach (22), zwiększone stężenie kwasu mlekowego (23), zmniejszenie przesączania kłębuszkowego (24), nasilenie przeciwtransportu sodowo-litowego (13), wzrost reabsorpcji zwrotnej sodu (25), insulinooporność (18) i leczenie hipotensyjne (26) to proponowane czynniki odpowiedzialne za spadek klirensu kwasu moczowego w nadciśnieniu tętniczym.
Najbardziej prawdopodobna jest hipoteza, która zakłada, że u podstaw hiperurykemii w nadciśnieniu tętniczym leżą zaburzenia hemodynamiczne funkcji nerek (27). Zgodnie z tą teorią w nadciśnieniu tętniczym dochodzi do wzrostu oporu obwodowego, a szczególnie oporu wewnątrznerkowego, który prowadzi do obniżenia nerkowego przepływu krwi, zwłaszcza przepływu korowego, wywołując zaburzenia w transporcie cewkowym kwasu moczowego. W konsekwencji klirens nerkowy kwasu moczowego ulega obniżeniu, a jego stężenie we krwi wzrasta.
W badaniach chorych z jednostronnym zwężeniem tętnicy nerkowej stwierdzono znaczny spadek klirensu kwasu moczowego po stronie niedokrwionej. Chirurgiczna rewaskularyzacja prowadziła do całkowitej normalizacji klirensu (21). Messerli i wsp. (28) wykazali u pacjentów z nadciśnieniem tętniczym granicznym oraz nadciśnieniem tętniczym utrwalonym ujemną korelację pomiędzy poziomem kwasu moczowego w surowicy a nerkowym przepływem krwi. Wspomniani autorzy stwierdzili istotnie wyższy opór wewnątrznerkowy u chorych z nadciśnieniem tętniczym i towarzyszącą hiperurykemią niż u pacjentów z prawidłowym poziomem kwasu moczowego.
Zmiany hemodynamiczne w nerkach, odpowiedzialne za częste występowanie hiperurykemii w nadciśnieniu tętniczym dotyczą mikrokrążenia. W przebiegu choroby nadciśnieniowej dochodzi w tętniczkach nerkowych do rozwoju procesu o typie stwardnienia „arteriolosclerosis” ( nephrosclerosis arteriolaris benigna), który początkowo prowadzi do spadku nerkowego przepływu krwi, a następnie do upośledzenia przesączania kłębuszkowego (29). Hollenberg i wsp. są zdania, że spadek nerkowego przepływu krwi spowodowany prawdopodobnie obkurczeniem tętniczek odprowadzających, występuje już we wczesnej fazie nadciśnienia tętniczego, a za wzmożone napięcie naczyń nerkowych odpowiedzialna jest zwiększona aktywność adrenergiczna oraz aktywacja układu renina-angiotensyna-aldosteron (30). Silne wazokonstrykcyjne działanie przejawia w tym mechanizmie angiotensyna II. Liczni autorzy wykazali w tej sytuacji upośledzoną naczyniorozkurczową czynność śródbłonka (31, 32, 33). Z czasem trwania nadciśnienia tętniczego dochodzi do zmian strukturalnych w małych tętnicach i tętniczkach. Następuje zwiększenie stosunku grubości ściany do światła naczynia (zmniejszenie światła naczynia i zwiększenie grubości jego ściany), co prowadzi do wzrostu oporu naczyniowego. Zjawisko to określane jest mianem eutroficznego remodelingu naczyniowego. Przy dłuższej ekspozycji na czynnik uszkadzający, jakim są: nadciśnienie, menopauza lub postępujący wiek i dodatkowo kwas moczowy w swej rozpuszczalnej postaci, dochodzi do nieodwracalnego przegrupowania elementów strukturalnych naczynia wokół zwężonego światła i trwałego upośledzenia czynności wazodylatacyjnej w odpowiedzi na bodźce i lokalne niedokrwienie. Wpływ prozapalny i proliferacyjny kwasu moczowego na mięśniówkę gładką naczyń prowadzi do dysfunkcji śródbłonka, co w konsekwencji przekłada się na jego udział w rozwoju powikłań nerkowych i sercowo-naczyniowych (34). Zmniejszenie perfuzji nerek u chorych z nadciśnieniem tętniczym nakłada się na zmiany postępujące z wiekiem i prowadzi do nasilenia produkcji kwasu moczowego dwoma drogami: poprzez zmniejszenie sekrecji oraz nasilenie jego syntezy wskutek wzmożenia ekspresji genu oksydazy ksantynowej (35).
Badania ostatnich lat pozwoliły wyjaśnić charakter zaburzeń w transporcie cewkowym kwasu moczowego występujących w nadciśnieniu tętniczym. Transport kwasu moczowego w nefronie jest zjawiskiem złożonym. Powszechnie akceptowany jest 4-fazowy model Sorensena i Levinsona (36) przedstawiony na rycinie 1. Zgodnie z nim kwas moczowy ulega całkowitej filtracji kłębuszkowej. Ładunek filtracyjny jest następnie niemal całkowicie wchłaniany zwrotnie. Etap ten nazywamy reabsorpcją przedsekrecyjną. W dalszej części nefronu kwas moczowy podlega sekrecji cewkowej w ilości wynoszącej ok. 40-50% ładunku filtrowanego. Wreszcie dystalnie do miejsca sekrecji zachodzi reabsorpcja posekrecyjna około 75-80% wydzielonej przez cewki ilości kwasu moczowego.
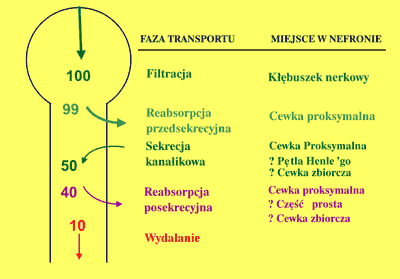
Ryc. 1. Transport kwasu moczowego w nefronie.
W nadciśnieniu tętniczym dochodzi właśnie do upośledzenia sekrecji kanalikowej kwasu moczowego z zachowaniem prawidłowej jego reabsorpcji przedsekrecyjnej i posekrecyjnej (36). Przyczyną obniżonej sekrecji kanalikowej kwasu moczowego w nadciśnieniu tętniczym jest zmniejszenie ładunku kwasu moczowego docierającego do przestrzeni okołocewkowej, a stąd do miejsc sekrecji w komórkach cewek nerkowych, na skutek obniżonego nerkowego przepływu krwi. W nadciśnieniu tętniczym stwierdzono dodatnią korelację pomiędzy klirensem i sekrecją kanalikową kwasu moczowego a nerkowym przepływem krwi (27).
Jednocześnie badania ostatnich lat wykazały zależność wydalania kwasu moczowego od natężenia przeciwtransportu sodowo-litowego w krwinkach czerwonych (23), reabsorpcji zwrotnej sodu (24), stopnia insulinooporności (25) i procesu starzenia, a więc czynników odgrywających rolę patogenetyczną w różnych postaciach nadciśnienia tętniczego. Najbardziej prawdopodobne mechanizmy rozwoju hiperurykemii w nadciśnieniu tętniczym przedstawia rycina 2.
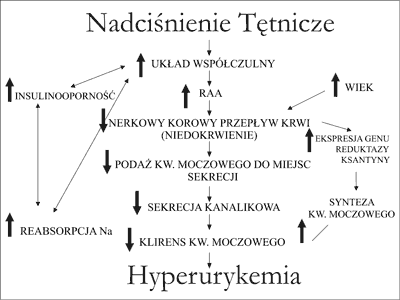
Ryc. 2. Mechanizm hiperurykemii w nadciśnieniu tętniczym.
Inne koncepcje związku hiperurykemii z nadciśnieniem tętniczym:
– Rozwój nefropatii moczanowej w przebiegu hiperurykemii prowadzi do nadciśnienia tętniczego (21).
– Zaburzenia czynności rdzenia nerek na skutek obecności złogów kwasu moczowego z odczynem zapalnym w tkance śródmiąższowej prowadzą do nadciśnienia tętniczego (8).
– Nadużywanie alkoholu jest wspólną przyczyną hiperurykemii i nadciśnienia tętniczego (37).
– W nadciśnieniu tętniczym ciężkim, z powikłaniami narządowymi, stwierdzono, obok redukcji wydalania, nasiloną produkcję kwasu moczowego, prawdopodobnie na skutek upośledzenia perfuzji tkankowej i niedotlenienia (38).
Kliniczne aspekty hiperurykemii
Coraz częściej donosi się, iż podwyższony poziom kwasu moczowego w surowicy stanowi niezależny czynnik ryzyka chorób układu sercowo-naczyniowego.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Jeżewska MM: Mechanizmy działania i rola oksydoreduktaz ksantynowych. Post Biochem 1974; 20: 259-279. Jeżewska MM. Xantine accumulation during hypoxantine oxidation by milk xantine oxidase. Eur J Biochem 1973; 36: 385-392.
2. Fox IH, Palella TD, Kelley WN: Hyperuricemia a marcer for cell energy crisis. N Engl J Med 1987; 317: 111-112.
3. Ward HJ: Uric acid as an independent risk factor in the treatment of hypertension. Lancet 1998; 352: 670-671.
4. Stanton JR, Freis ED: Serum uric acid concentration in essential hypertension. Proc. Soc. Exp. Biol. Med. 1947, 66, 193-200.
5. Duncan H: Gout, familiar hyperuricemia and renal disease. Quart. J. Med. 1960, 113, 127-129.
6. Breckenridge A: Hypertension and hyperuricemia. Lancet, 1966, 1, 15-18.
7. Bulpitt CJ: Serum uric acid in hypertensive patients. Br. Heart J. 1975, 37, 1210-1215.
8. Cannon PJ et al.: Hyperuricemia in primary and renal hypertension. N. Engl. J. Med. 1966, 275, 457-462.
9. Grajek S i wsp.: Nadciśnienie tętnicze u mężczyzn w wieku 40-59 lat. Cz.III. Średnie stężenie kwasu moczowego w surowicy hiper- i normotoników. Zależność kwasu moczowego od ciśnienia tętniczego, czasu trwania choroby, wieku, cholesterolu i ciężaru ciała. Kard. Pol. 1983, 26, 149-154.
10. Schrade W, Bochle E, Biegler R: Humoral changes in atherosclerosis: investigation on lipids, fatty acids, ketones bodies, pyruvic acid and glucose in blood. Lancet, 1960, 2, 409-412.
11. Grayzel AI, Liddle L, Seegmiller JE: Diagnostic significance of hyperuricemia in arthritis. N. Engl. J. Med. 1961, 265, 763-769.
12. Itskowitz HD, Sellers A: Gout and hyperurycemia after adrenalectomy for hypertension. N. Engl. J. Med. 1963, 268, 1105-110.
13. Kanbay M et al.: Effect of treatment of hyperuricemia with allopurinol on blood pressure, creatinine clearence, and proteinuria in patients with normal renal functions. Int Urol Nephrol 2007; 39: 1227-1233.
14. Franco MCP et al.: Effects of Low Birth Weight in 8- to 13-Year-Old Children. Implications in Endothelial Function and Uric Acid Levels. Hypertension 2006; 48: 45-50.
15. Krishnan E et al.: Hyperuricemia and Incidence of Hypertension Among Men Without Metabolic Syndrome. Hypertension 2007; 49: 298-303.
16. Lee JE KY et al.: Serum Uric Acid Is Associated With Microalbuminuria in Prehypertension. Hypertension. 2006; 47: 1-6.
17. Mazzali M et al.: Elevated uric acid increases blood pressure in the rat by a novel crystal-dependent mechanism. Hypertens 2001; 38: 1101-1106.
18. Facchini F et al.: Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. JAMA 1991; 266: 3008-3015.
19. Culleton BF et al.: Serum uric acid and risk for cardiovascular disease and death: the Framingham Heart Study. Ann Intern Med 1999; 131: 7-13.
20. Yoo TW et al.: Relationship between serum uric acid concentration and insulin resistance and metabolic syndrome. Circ J 2005; 69: 928-33.
21. Simon NM et al.: Differential uric acid excretion in essential and renal hypertension. Circulation, 1969, 39, 121-128.
22. Editorial: Hypertension and uric acid. Lancet, 1981, 1, 365-368.
23. Demartini FE et al.: Lactic acid metabolism in hypertensive patients. Science, 1965, 148, 1482-1488.
24. Strazzullo P, Cappuccio FP, Trevisan M: Red blood cell sodium-lithium countertransport, blood pressure and uric acid metabolism in untreated healthy men. Am. J. Hypertension, 1989, 2, 634-638.
25. Cappuccio P, Farinaro E, Trevisan M: Uric acid metabolism and tubular sodium handling. JAMA, 1993, 3, 270-276.
26. Barsotti G et al.: Serum uric acid in mild essential hypertension. Clin. Nephrol. 1983, 45, 145-149.
27. Tykarski A: Evaluation of renal handling of uric acid in essential hypertension: hyperuricaemia related to decreased urate secretion. Nephron, 1991, 59, 364-368.
28. Messerli FH et al.: Serum uric acid in essential hypertension: An indicator of renal vascular involvement. Ann. Intern. Med. 1980, 23, 817-821.
29. Johnson RJ et al.: A unifying pathway for essential hypertension. Am J Hypertens 2005,18: 431-440.
30. Hollenberg NK, Borucki LJ, Adams DF: The renal vasculature in early essential hypertension: evidence for pathogenetic role. Medicine 1978; 57: 167-176.
31. Panza AJ et al.: Role of endothelium-derived nitric oxide in the abnormal endothelium-dependent vascular relaxation of patients with essential hypertension. Circulation 1993; 87: 1468-1474.
32. Panza JA et al.: Impaired endothelium-dependent vasodilation in patients with essential hypertension. Evidence that nitric oxide abnormality is not localized to a single signal transduction pathway. Circulation 1995; 91: 1732-1738.
33. Salazar FJ et al.: Renal effects of prolonged synthesis inhibition of endothelium-derived nitric oxide. Hypertension 1992; 20: 113-117.
34. Sanchez-Lozada LG et al.: Hormonal and cytokine effects of uric acid. Curr O Nephr Hypertens. 2006, 15(1): 30-33.
35. Berry CE, Hare JM: Xanthine oxidoreductase and cardiovascular disease – molecular mechanisms and pathophysiologic implications. J Physiol 2004; 555(Part 3): 589-606.
36. Levinson DJ, Sorensen LB: Renal handling of uric acid in normal and gouty subjects: evidence for a 4-component system. Ann. Rheum. Dis. 1980, 39, 173-179.
37. Ramsey LE: Hyperuricemia in hypertension: role of alcohol. Br. Med. J. 1979, 1, 653-659.
38. Łopatka P: Mechanizm hiperurykemii w nadciśnieniu tętniczym ciężkim i opornym na leczenie. Praca doktorska. Akademia Medyczna w Poznaniu, Poznań 2000.
39. Bickel Ch et al.: Serum uric acid as an independent predictor of mortality in patients with angiographically proven coronary artery disease. Am J Cardiol 2002; 89: 12-17
40. Struthers AD et al.: Effect of allopurinol on mortality and hospitalizations in chronic heart failure: a retrospective cohort study. Heart 2002; 87: 229-234.
41. Levine Wet al.: Serum uric acid and 11.5-year mortality of middle-aged women: findings of the Chicago Heart Association Detection Project in Industry. J Clin Epidemiol 1989; 42: 257-267.
42. Freedman DS et al.: Relation of serum uric acid to mortality and ischemic heart disease: The NHANES I epidemiologic follow-up study. Am J Epidemiol 1995; 141: 637-644.
43. Bengtsson C et al.: Hyperuricaemia and risk of cardiovascular disease and overall death: a 12-year follow-up of participants in the population study of women in Gothenburg, Sweden. Acta Med Scand 1988; 224: 549-555.
44. Fang J, Alderman MH: Serum uric acid and cardiovascular mortality. The NHANES I epidemiologic follow up study, 1971-1992: National Health and Nutrition Examination Survey. JAMA 2000; 283: 2404-2410.
45. Alderman MH: Uric acid and cardiovascular risk. Curr Opin Pharmacol 2002; 2: 126-130.
46. Tykarski A: Zależność pomiędzy stężeniem kwasu moczowego w surowicy a zaawansowaniem zmian naczyniowych i przerostu mięśnia sercowego w nadciśnieniu tętniczym pierwotnym. Pol. Arch. Med. Wewn. 1991, 86, 183-189.
47. Ostrander LD, Lamphiear DE: Coronary risk factors in a community. Circulation, 1976, 53, 152-156.
48. Coronary Drug Project Research Group: Serum uric acid: Its association with other risk factors and with mortality in coronary heart disease. J. Chronic Dis. 1976, 29, 557-561.
49. Klein R et al.: Serum uric acid. Its relationship to coronary heart disease risk factors and cardiovascular disease. Arch. Intern. Med. 1973, 132, 401-409.
50. Hall AP: Correlation among hyperuricaemia, hypercholesterolemia, coronary disease and hypertension. Arth. Rheum. 1964, 8, 846-852.
51. Benedek TG: Correlation of serum uric acid and lipid concentration in normal, gouty and atherosclerotic men. Ann. Intern. Med., 1967, 66, 85-91.
52. Chobanian AV et al.: The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: the JNC 7 report. JAMA 2003; 289: 2560-2572.
53. Scott J, Sturge RA: The effects of weight loss on plasma and urinary uric acid and lipid levels. Adv. Exp. Med. Biol. 1977, 76B, 274-277.
54. Berry C et al.: Investigation into the sources of superoxide in human blood vessels: angiotensin II increases superoxide production in human internal mammary arteries. Circulation 2000; 101: 2206-2212.
55. Reuben GM, Vanhoutte PM: Superoxide anion and hypoxia inactivate endothelium-derived relaxing factor. Am J Physiol 1986; 250: H 822-827.
56. Mustard JF, Murphy EA, Ogryrlo MA: Blood coagulation and platelet economy in subjects with primary gout. Can. Med. Assoc. J. 1963, 89, 1207-1212.
57. Ginsberg MH, Kozin F, McCarty DJ: Release of platelet constituents by monosodium urate crystals. J. Clin. Invest. 1977, 60, 999-1005.
58. Godin M: Hypertension and hyperuricemia: quelle relations? Nouv. Presse Med. 1982, II, 643-649.
59. Fridovich I: Quantitative aspects of the production of superoxide anion radical by milk xanthine oxidase. J. Biol. Chem. 1970, 245, 4053-4059.
60. Becker BF, Reinholz N: Uric acid as radical scavenger and antioxidant in the heart. Pfligers Arch. 1989, 415, 127-132.
61. Wong KYK: Urate predicts subsequent cardiac death in stroke survivors. Eur. Heart J. 2002, 23, 788-793.
62. Anker SD et al.: Uric acid and survival in chronic heart failure. Validation and application in metabolic, functional, and hemodynamic staging. Circulation 2003; 107: 1991-1997.
63. Hare JM et al.: Impact of Oxypurinol in Patients With Symptomatic Heart Failure: Results of the OPT-CHF Study. J Am Coll Cardiol 2008; 51: 2301-2309.
