© Borgis - Postępy Nauk Medycznych 4/2011, s. 260-266
*Jan Kuś
Zwykłe śródmiąższowe zapalenie płuc, czyli samoistne włóknienie płuc
Usual interstitial pneumonia/idiopathic pulmonary fibrosis
I Klinika Chorób Płuc Instytutu Gruźlicy i Chorób Płuc w Warszawie
Kierownik Kliniki: prof. dr hab. med. Jan Kuś
Streszczenie
Zwykłe śródmiąższowe zapalenie płuc (UIP) jest określeniem zmian stwierdzanych w badaniu mikroskopowym wycinka płuca pobranego od chorych z klinicznym obrazem samoistnego włóknienia płuc (SWP). Samoistne włóknienie płuc jest chorobą postępującą, która nie poddaje się leczeniu. Średnie przeżycie chorych od chwili rozpoznania wynosi 2-3 lata i jest podobne jak w raku płuca. Rozpoznanie SWP ustala się na podstawie objawów klinicznych, badań radiologicznych i biopsji płuca. Kilka lat temu opublikowano wytyczne międzynarodowej grupy ekspertów, w których podane są kryteria rozpoznania tej choroby bez wykonywania biopsji płuca. Nie ma skutecznego leczenia farmakologicznego SWP. U części chorych, zgodnie z międzynarodowymi wytycznymi, należy podjąć próbę leczenia immunosupresyjnego, które jednak często jest nieskuteczne. Wybrani chorzy są kwalifikowani do przeszczepienia płuc. W pozostałych przypadkach stosuje się leczenie objawowe, a w okresie niewydolności oddychania tlenoterapię domową.
Summary
Usual interstitial pneumonia (UIP) is a pathological pattern found in microscopic examination of lung tissue obtained by lung biopsy from patients presenting symptoms of idiopathic pulmonary fibrosis (IPF). Idiopathic pulmonary fibrosis is progressive disease of the lung for which no effective treatment exist. The mean survival is 2-3 years which is similar to mean survival of lung cancer. The diagnosis of IPF is determined by clinical symptoms, radiographic images and lung biopsy. There are international guidelines which define criteria of the diagnosis of IPF without lung biopsy. In the selected cases the immunosuppressive treatment according to the international guidelines should be tried. The pharmacologic treatment is often ineffective and lung transplantation should be considered in selected cases. Best supportive care including home oxygen therapy is the treatment to offer in the advanced stage of the disease.

Samoistne włóknienie płuc (SWP) jest jedną z postaci idiopatycznych zapaleń śródmiąższowych płuc. Jego podstawową cechą jest przewlekłe, postępujące włóknienie płuc, którego obraz w badaniu mikroskopowym wycinka z płuc odpowiada zwykłemu śródmiąższowemu zapaleniu, zwanemu w skrócie UIP od angielskiej nazwy usual interstitial pneumonia (1). Etiologia samoistnego włóknienia płuc jest nieznana. Przypuszcza się, że jest to proces nieprawidłowego gojenia wielokrotnych, mikroskopijnych uszkodzeń nabłonka pęcherzyków płucnych przez nieokreślone bliżej czynniki z tworzeniem się ognisk fibroblastów i miofibroblastów (2). W międzynarodowej klasyfikacji chorób śródmiąższowych zaproponowanej przez ekspertów American Thoracic Society (ATS) i European Respiratory Society (ERS) zwykłe śródmiąższowe zapalenie płuc (UIP) jest wyodrębnione spośród pozostałych zapaleń śródmiąższowych innych niż UIP (non-UIP) ze względu na to, że UIP wyróżnia się największą częstością występowania spośród idiopatycznych zapaleń śródmiąższowych, bardzo złym rokowaniem i brakiem skutecznego leczenia (3). UIP jest określeniem zmian mikroskopowych u chorych z klinicznym rozpoznaniem samoistnego włóknienia płuc (SWP). W SWP nigdy nie obserwuje się samoistnych remisji, a średnie przeżycie w tej chorobie jest porównywalne do przeżycia w chorobach nowotworowych i wynosi od chwili rozpoznania 2-3 lata (4). Choroba występuje zwykle u osób po 50. roku życia, częściej u mężczyzn. Zachorowalność jest bardzo niska przed 60. rokiem życia, wynosi poniżej 2/100 000 i gwałtownie wzrasta z wiekiem do około 11/100 000 w grupie wieku 60-69 lat i powyżej 20/100 000 w grupie wieku 70-79 lat (4, 5). Początek choroby jest podstępny, często trudno uchwytny. Chorzy zgłaszają suchy kaszel i duszność po wysiłku nasilającą się od miesięcy, a nawet lat. W badaniu fizykalnym należy zwrócić uwagę na palce pałeczkowate, które występują u ponad połowy chorych. Osłuchiwaniem płuc stwierdza się dyskretne trzeszczenia u podstawy obu płuc, najlepiej słyszalne pod koniec wdechu. Typowe dla SWP trzeszczenia słyszalne na szczycie wdechu nad dolnymi polami płuc bywają przez lekarza pierwszego kontaktu interpretowane jako związane z niewydolnością krążenia lub zapaleniem płuc.
W badaniach czynnościowych płuc typowe są cechy restrykcji, czyli obniżenie pojemności życiowej – VC (vital capacity) i całkowitej pojemności płuc – TLC (total lung capacity). Obniżona pojemność życiowa i dobrze zachowana drożność oskrzeli, duża w stosunku do objętości płuc, powodują, że w badaniu spirometrycznym stwierdza się często stosunek objętości wydechowej pierwszosekundowej (FEV1) do pojemności życiowej (VC), czyli wskaźnik FEV1/VC wyższy od spodziewanego. We wczesnych stadiach choroby, kiedy zmiany w płucach są minimalne, podstawowe badanie czynnościowe, jakim jest spirometria, może nie wykazywać zaburzeń restrykcyjnych (6). Włóknienie płuc powoduje zaburzenie dyfuzji tlenu z powietrza w pęcherzykach płucnych do krwi włośniczek oplatających pęcherzyki. W praktyce do pomiaru tego zaburzenia stosuje się pomiar zdolności dyfuzyjnej dla tlenku węgla (DLCO). Jest to czuły wskaźnik zaburzeń czynnościowych w samoistnym włóknieniu płuc, który wypada nieprawidłowo wcześniej niż widoczne są zmiany w badaniu spirometrycznym (7). Podobnie, wyniki badania gazów krwi w spoczynku mogą być również prawidłowe. Należy wtedy wykonać test wysiłkowy, który wykaże, że po wysiłku ujawnia się hipoksemia.
Na konwencjonalnym radiogramie klatki piersiowej zwraca uwagę wyższe ustawienie obu kopuł przepony oraz zmniejszona wielkość pól płucnych. W okolicach nadprzeponowych widoczne są zacienienia o typie siateczki. W bardziej zawansowanym stadium zacienienia sięgają coraz wyżej, ale zawsze są najbardziej nasilone w obwodowych częściach płuc, czyli pod opłucną.
U chorych z podejrzeniem samoistnego włóknienia płuc nieodzowna jest tomografia komputerowa płuc o wysokiej rozdzielczości (TKWR). Zmiany, które można uwidocznić przy pomocy tej metody, są bardzo charakterystyczne. Są to zacienienia o wyglądzie siateczki oraz plastra miodu najbardziej nasilone w dolnych częściach płuc. Jest ich więcej z tyłu niż z przodu. Obszary zacienień typu matowej szyby nie występują lub są niewielkie. Nie występują guzki. Ten obraz jest uważany za typowy dla samoistnego włóknienia płuc i pozwala na rozpoznanie choroby bez wykonywania biopsji płuca z ryzykiem błędu mniejszym niż 10% (8).
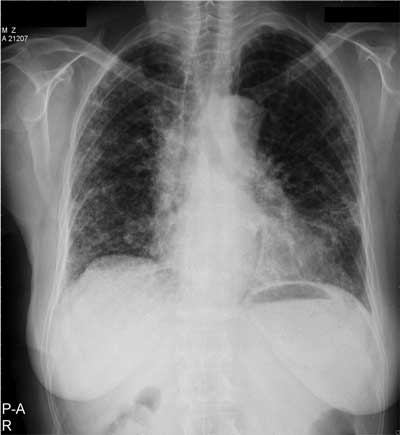
Ryc. 1 a. Chora lat 64, od kilku lat odczuwa stopniowo narastającą duszność wysiłkową. Radiogram przeglądowy płuc. Pola płucne są małe, przepona ustawiona wysoko. W obu polach płucnych widoczne rozsiane zmiany typu siateczki, najbardziej nasilone w dolnych polach płuc i pod opłucną. Obraz wskazuje na samoistne włóknienie płuc.
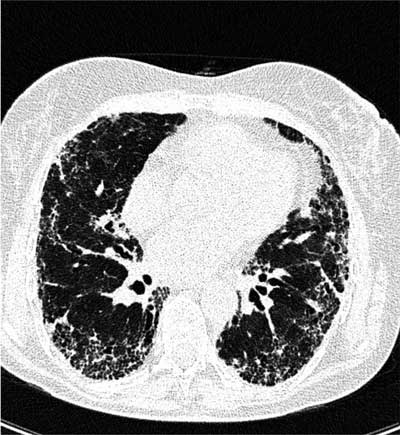
Ryc. 1 b. Tomografia komputerowa wysokiej rozdzielczości płuc chorej z ryciny 1 a. W obu płucach zacienienia typu plastra miodu zlokalizowane pod opłucną. Obraz odpowiada zwykłemu, śródmiąższowemu zapaleniu płuc (UIP).
Jak wspomniano na wstępie, samoistnemu włóknieniu płuc odpowiada obraz mikroskopowy wycinka płuca określany jako zwykłe śródmiąższowe zapalenie płuc – UIP. Za typowe cechy UIP w badaniu mikroskopowym uważa się mozaikowatość zmian, czyli występowanie obszarów prawidłowego miąższu płuc na przemian z obszarami zmienionymi zapalnie, zaburzenie prawidłowej architektury płuca przez blizny i obszary plastra miodu oraz różnoczasowość zmian (9). Zmiany są określane jako różnoczasowe z powodu występowania obok siebie obszarów „starych” zmian w postaci dokonanego włóknienia i „świeżych” w postaci skąpych nacieków zapalnych (10). Nigdy jednak nie wykazano, aby najpierw występowały większe nacieki zapalne, a potem włóknienie, stąd przypuszcza się, że obydwa typy zmian występują równocześnie od początku choroby. Wśród opisanych zmian występują depozyty kolagenu oraz skupienia fibroblastów rozmieszczone głównie na obrzeżach ognisk bliznowacenia. Im więcej jest widocznych skupisk fibroblastów, tym gorsze jest rokowanie chorego co do życia (11). Rozstrzenie drobnych oskrzeli i oskrzelików z pociągania tworzą cysty, które dają mikroskopowy obraz plastra miodu. Nacieki zapalne z komórek jednojądrowych nie występują lub są bardzo niewielkie. Nie występują ziarniniaki i depozyty substancji mineralnych, jak włókna azbestu czy kryształy krzemionki. Podobnie jak w badaniu radiologicznym, również w badaniu mikroskopowym zmiany są najbardziej nasilone w dolnych częściach płuc i pod opłucną (3).
Chorzy podejrzani o SWP powinni mieć wykonaną bronchoskopię z płukaniem oskrzelowo-pęcherzykowym – BAL (broncho-alveolar-lavage) i przezoskrzelową biopsję płuca. Celem tych badań jest wykluczenie innej choroby śródmiąższowej, infekcyjnej i zmian rozrostowych. Skład komórek w płynie z płukania oskrzelowo-pęcherzykowego nie ma cech szczególnych wskazujących na SWP, ale podwyższony ponad normę odsetek limfocytów przemawia przeciwko tej chorobie (12). Pobrana w czasie bronchoskopii biopsja przezoskrzelowa daje bardzo mały wycinek płuca, który nie pozwala na rozpoznanie SWP, ale służy do wykluczenia choroby ziarniniakowej, infekcyjnej i raka pęcherzykowo-oskrzelikowego, który w obrazie radiologicznym przypomina chorobę śródmiąższową.
We wspomnianych wyżej wytycznych ATS/ERS odnośnie postępowania w śródmiąższowych zapaleniach płuc podano kryteria rozpoznania samoistnego włóknienia płuc bez wykonywania chirurgicznej biopsji płuca. Wyodrębniono duże i małe kryteria, które są następujące (3).
Kryteria duże:
1. Wykluczono znane przyczyny choroby śródmiąższowej, jak reakcje na leki, czynniki środowiska i choroby tkanki łącznej.
2. Nieprawidłowe wyniki badań czynnościowych płuc wykazują restrykcję (obniżenie VC, często ze wzrostem wskaźnika FEV1/FVC oraz upośledzenie wymiany gazowej (wzrost P[A-a]O2*, obniżenie PaO2 w spoczynku lub po wysiłku i obniżenie DLCO**).
3. W badaniu płuc metodą tomografii komputerowej wysokiej rozdzielczości TKWR widoczne są w częściach przypodstawnych zacieniena o typie siateczki z minimalną komponentą zmian typu matowej szyby.
4. Biopsja przezoskrzelowa lub BAL*** nie wskazują na inne rozpoznanie.
Kryteria małe:
1. Wiek > 50 lat.
2. Powolny (podstępny) początek narastania duszności wysiłkowej.
3. Utrzymywanie się objawów od ponad 3 miesięcy.
4. Obustronne trzeszczenia słyszalne na wdechu nad dolnymi polami płuc.
*P[A-a]O2 – różnica ciśnienia parcjalnego tlenu między powietrzem w pęcherzykach płuc a krwią tętniczą.
**DLCO – zdolność dyfuzyjna dla tlenku węgla.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. King TE Jr: Idiopathic pulmonary fibrosis. [In:] Schwarz M, King TE (ed.): Interstitial lung disese. Fifth ed. Shelton, Connecticut, USA. People’s Medical Publishing House – USA 2011; 895-944.
2. Selman M, King TE Jr, Pardo A: Idiopathic pulmonary fibrosis: prevailing and evolving hypothesis about its pathogenesis and implications for therapy. Ann Intern Med 2001; 134: 136-151.
3. American Thoracic Society/European Respiratory Society: international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2002; 165: 277-304.
4. Fernandez Perez ER et al.: Incidence, prevalence and clinical course of idiopathic pulmonary fibrosis. A population based study. Chest 2010; 137, 1: 29-137.
5. Coultas DB et al.: The epidemilogy of interstitial lun diseases. Am J Respir Cit Care Dis 1994; 150: 967-972.
6. Kowalski J, Radwan L, Boros P: Zaburzenia czynności układu oddechowego w chorobach śródmiąższowych płuc (ChŚP). [W:] Kowalski J, Koziorowski A, Radwan L (ed.): Ocena czynności płuc w chorobach układu oddechowego. Borgis Wydawnictwo Medyczne Warszawa, 2004; 120-128.
7. Du Bois RM: Cryptogenic fibrosing alveolitis. [In:] Warrell DA, Cox TM, Firth JD, Benz EJ (ed.): Oxford textbook of medicine. Fourth edition. New York, Oxford University Press Inc 2005; vol. 2: 1446-1452.
8. Hunninghake GW, Lynch DA, Calvin JR et al.: Radiologic findings are strongly associated with a pathologic diagnosis of usual interstitial pneumonia. Chest 2005; 124: 1215-1223.
9. Katzenstein A-LA, Mukhopadhyay S, Myers JL: Diagnosis of usual interstitial pneumonia and distinction from other fibrosing interstitial lung diseases. Human Pathol 2008; 39: 1275-1294.
10. Myers JL, Katzenstein ALA: Beyond a consensus classification for idiopathic interstitial pneumonias: progress and controversies. Histopathology 2009; 54: 90-103.
11. Tiitto L, Bloigu R, Heiskanen U et al.: Relationship between histopathological features and the course of idiopathic pulmonary fibrosis/usual interstitial pneumonia. Thorax 2006; 61: 1091-1095.
12. Nagai S, Hanha T, Ito Y et al.: Bronchoalveolar lavage in idiopathic interstitial lung diseases. Semin Respi Crit Care Med 2007; 28: 496-503.
13. Patel NM, Lederer DJ, Borczuk AC et al.: Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest 2007; 132: 998-1006.
14. Corte TJ, Wort SJ, Gatzoulis MA et al.: Elevated brain naturiuretic peptide predicts mortality in interstitial lung disease. Eur Respir J 2010; 36: 819-825.
15. Lettieri ChJ, NathanSD, Barnett SD et al.: Prevalance and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest 2006; 129: 746-752.
16. American Thoracic Society: Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. Am J Respir Crit Care Med 2000; 161: 646-664.
17. BTS: Interstitial lung disease guideline. Thorax 2008; 63: v1-v58.
18. Demedts M, Behr J, Buhl R et al.: High dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2005; 353: 2229-2242.
19. Maher TM, Wells AU: Optimal treatment for idiopathic pulmonary fibrosis. Thorax 2008; 63: 1120-1121.
20. Noth I, Martinez FJ: Recent advances in idiopathic pulmonary fibrosis. Chest 2007; 132: 637-650.
21. Oku H, Shimizu T, Kawabata T et al.: Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokinez and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur J Pharmacol 2008; 590: 400-408.
22. Azuma AP: antifibrotic agent for idiopathic pulmonary fibrosis. Expert Rev Respir Med 2010; 4: 301-310.
23. Azuma A et al.: Double-blind placebo controled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J respir crit Care Med 2005; 171: 1040-1047.
24. Kim DS, Collard HR, King TE Jr: Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc 2006; 3: 285-292.
25. Raghu G et al.: Idiopathic pulmonary fibrosis study group. A placebo kontroled trial: of interferon-gamma 1b in patients with idiopathic pulmonary fibrosis. N Engl J Med 2004; 350: 125-133.
26. Song JW, Hong S-B, Lim C-M et al.: Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011; 37: 356-363.
27. Kim DS et al.: Acute exacerbation of idiopathic pulmonary fibrosis: frequency and clinical features. Eur respir j 2006; 27: 143-150.
28. Agarwal R, Jindal SK: Acute exacerbation of idiopathic pulmonary fibrosis: a systematic review. Eur J Intern Med 2008; 19: 227-235.
29. Parambil JG, Myers JL, Ryu JH: Histopathologic features and outcome of patients with acute exacerbation of idiopathic pulmonary fibrosis undergoing surgical lung biopsy. Chest 2005; 128: 3310-3315.
30. Kus J: Współistnienie samoistnego włóknienia płuc i rozedmy. Pneumonol Alergol Pol 2009; 77: 115-117.
31. Cottin V, Nines H, Delaval P-Y et al.: Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur respir J 2005; 26: 586-593.
32. Niewoehner DE, Hoidal JR: Lung fibrosis and emphysema; divergent responses to common injury? Science 1982; 217: 359-360.
33. Collard HR, Ryu JH, Douglas WW et al.: Combined corticosteroid and cyclophosphamide therapy does not alter survival in idiopathic pulmonary fibrosis. Chest 2004; 125: 2169-2174.
