© Borgis - Postępy Nauk Medycznych 4/2011, s. 324-331
*Małgorzata Bartosiewicz
Zmiany śródmiąższowe w płucach w przebiegu twardziny układowej, chorób zapalnych mięśni, zespołu Sjögrena oraz mieszanej choroby tkanki łącznej
Interstitial lung disease in systemic sclerosis, idiopathic inflamatory myopathies, Sjögrens syndrome and mixed connective tissue diseases
I Klinika Chorób Płuc Instytutu Gruźlicy i Chorób Płuc w Warszawie
Kierownik Kliniki: prof. dr hab. med. Jan Kuś
Streszczenie
Choroby układowe tkanki łącznej to niejednorodna grupa schorzeń o podłożu autoimmunologicznym w przebiegu których dochodzi do zajęcia wielu układów i narządów. Choroby śródmiąższowe płuc są dobrze znaną manifestacją chorób układowych. Zajęcie układu oddechowego stanowi istotną przyczynę zwiększonej chorobowości i śmiertelności w chorobach układowych. Częstość występowania, objawy kliniczne, rokowanie oraz odpowiedź na leczenie zależą od typu zmian w płucach oraz rodzaju choroby zasadniczej. Powikłania płucne w przebiegu twardziny układowej są bardzo częste i obecnie stanowią główną przyczynę zgonów. Na podstawie badań autopsyjnych wykazano, że występują one u większości pacjentów. Choroba śródmiąższowa płuc jest również dobrze znanym powikłaniem zapalenia wielomięśniowego i skórnomięśniowego, ale częstość jej wysterowania i naturalny przebieg wciąż pozostają nie do końca wyjaśnione. Zaburzenia czynności układu oddechowego są często opisywanym pozagruczolowym powikłaniem zespołu Sjögrena i mieszanej choroby tkanki łącznej. W poniższym artykule dokonano przeglądu zmian śródmiąższowych w płucach towarzyszących twardzinie układowej, miopatiom zapalnym, zespołowi Sjögrena oraz mieszanej chorobie tkanki łącznej.
Summary
The connective tissue diseases are a variable group of autoimmune mediated disorders characterized by multiorgan damage. Interstitial lung disease is a well-recognized manifestation of connective tissue disease. Lung involvement is an increasing cause of mortality and morbidity in the connective tissue diseases. The frequency, clinical presentation, prognosis and response to therapy are different, depending on the pattern of involvement as well as on the underlying connective tissue disease. Pulmonary involvement in systemic sclerosis is very common and became the first cause of death in this disease. Autopsy studies have documented lung involvement in majority of patients. Interstitial lung disease is also well-recognized manifestation of polymyositis and dermatomyositis but its prevalance and natural course are poorly understood. Pulmonary abnormalities are often described extraglandular systemic manifestation of Sjögrens syndrome and mixed connective tissue disease. In this article, interstitial lung disease accompanying systemic sclerosis, idiopathic inflammatory myopathies, Sjögrens syndrome and mixed connective tissue disease are reviewed.

Do rzadziej występujących układowych chorób tkanki łącznej należą: twardzina układowa, choroby zapalne mięśni – zapalenie wielomięśniowe i skórnomięśniowe, zespół Sjögrena i mieszana choroba tkanki łącznej. W ich przebiegu, podobnie jak w reumatoidalnym zapaleniu stawów i toczniu rumieniowatym układowym, może dochodzić do rozwoju zmian śródmiąższowych w płucach, które są wynikiem toczącego się w nich procesu immunologicznego. Chorobą, w której zmiany śródmiąższowe w płucach spotyka się najczęściej, jest twardzina układowa. Objawy ze strony układu oddechowego mogą wyprzedzać, czasami nawet na wiele lat, inne typowe dla kolagenoz objawy. Poniżej przedstawiona zostanie charakterystyka zmian śródmiąższowych w płucach u chorych na twardzinę układową, miopatie zapalne, zespół Sjögrena i mieszaną chorobę tkanki łącznej.
Twardzina układowa (SSc – systemic sclerosis)
Twardzina układowa jest przewlekłą układową chorobą tkanki łącznej, dotycząca głównie kobiet w wieku 45-64 lata, w której współistnieją procesy zapalenia, vaskulopatii i włóknienia, które doprowadzają do zaburzeń funkcji i niewydolności wielu narządów wewnętrznych, w tym układu oddechowego, pokarmowego, krążenia i nerek (1). Przebieg kliniczny choroby jest zróżnicowany. Wyróżnia się 2 główne postacie choroby: postać uogólnioną i ograniczoną. W postaci uogólnionej (30% przypadków) dochodzi do zajęcia proksymalnych i dystalnych odcinków skóry, przebieg choroby jest dynamiczny i już we wczesnym okresie występują poważne powikłania narządowe. U 30% chorych obecne są przeciwciała przeciwko topoizomerazie I (Scl-70), a przeciwciała antycentromerowe (ACA) zwykle są nieobecne, stwierdza się je jedynie u 3% pacjentów (2). Wykazano dodatnią korelację pomiędzy wielkością FVC (natężona pojemność życiowa – forced vital capacity), zmianami w obrazie TKWR płuc (tomografia komputerowa wysokiej rozdzielczości) oraz poziomem przeciwciał Scl-70, które mogą być markerem choroby śródmiąższowej płuc w uogólnionej postaci twardziny układowej (3). W postaci ograniczonej (70% przypadków) choroba ma przebieg przewlekły, postępujący, z wieloletnim wywiadem objawu Raynauda, pozytywnymi przeciwciałami antycentromerowymi u 70% chorych, zmiany skórne dotyczą dystalnych odcinków kończyn i twarzy, powikłania narządowe występują później, generalnie rokowanie jest lepsze (2, 4, 5). Nadciśnienie płucne i choroba śródmiąższowa płuc należą do najczęstszych i najbardziej istotnych manifestacji płucnych twardziny układowej.
Klinicznie jawna choroba śródmiąższowa płuc stwierdzana jest u ok. 40% chorych (6) i stanowi jedną z głównych przyczyn zgonów (ok. 30%) pacjentów chorych na sklerodermię (7). W badaniach autopsyjnych cechy choroby śródmiąższowej płuc widoczne są u 75% chorych (4). U 13-15% chorych w przebiegu choroby dochodzi do rozwoju zaawansowanego włóknienia płuc i ciężkich zaburzeń restrykcyjnych stwierdzanych w badaniach czynnościowych układu oddechowego (5, 8). Do rozwoju choroby śródmiąższowej płuc predysponuje postać uogólniona twardziny, ale może się ona rozwinąć u pacjentów z postacią ograniczoną choroby, jak również u chorych bez zmian skórnych (6, 9). Choroba śródmiąższowa płuc o gwałtownym przebiegu zwykle rozwija się w ciągu pierwszych 2-4 lat trwania choroby (5, 6, 9).
Zaburzenia motoryki przełyku (w 80% przypadków bezobjawowe) i związane z tym wielokrotne aspiracje kwaśnej treści żołądkowej do układu oddechowego sprzyjają rozwojowi zmian śródmiąższowych (6, 10). Objawy kliniczne choroby śródmiąższowej płuc są niespecyficzne – najczęściej chorzy zgłaszają kaszel i postępującą duszność wysiłkową, a w badaniu przedmiotowym w przypodstawnych częściach płuc obecne są trzeszczenia. Złotym standardem w rozpoznawaniu zmian śródmiąższowych w płucach jest tomografia komputerowa wysokiej rozdzielczości (ryc. 1, 2).
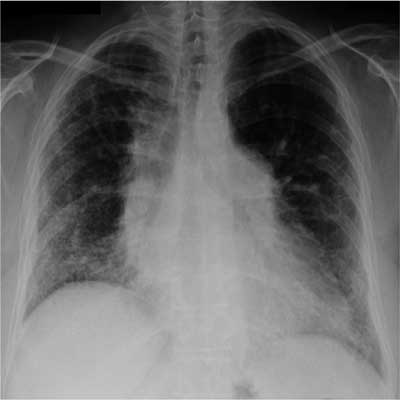
Ryc. 1. Zdjęcie radiologiczne klatki piersiowej – twardzina układowa (Scl-70+++). Zmniejszona objętość pól płucnych, w obu płucach zmiany rozsiane o charakterze siateczkowatym, głównie w obwodowych partiach płuc.
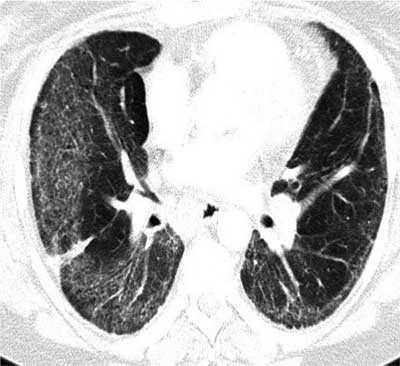
Ryc. 2. TKWR płuc – twardzina układowa (Scl-70+++). W obu płucach obecne zmiany rozsiane o charakterze śródmiąższowym zlokalizowane obwodowo-pogrubiałe struktury śródmiąższu tworzą obraz dość drobnej siatki, na ich tle widoczne są rozstrzenie oskrzeli z pociągania.
Na podstawie analizy wyników randomizowanego wieloośrodkowego, kontrolowanego placebo badania Scleroderma Lung Study (SLS) oraz wyników badań retrospektywnych dotyczących chorych z chorobą śródmiąższową płuc w przebiegu twardziny zaproponowano system kwalifikacji chorych oparty na wyniku badania TKWR płuc pomocny w podejmowaniu decyzji terapeutycznych (11). Pacjenci, u których zmiany w płucach dotyczą > 20% obszaru płuc w TKWR, mają istotnie gorsze rokowanie (choroba poważna), pacjenci ze zmianami dotyczącymi < 10% obszaru płuc mają rokowanie dobre (choroba łagodna), a u chorych ze zmianami pośrednimi (> 10 i < 20%) decydujące znaczenie mają wyniki badań czynnościowych układu oddechowego – w tym wartość FVC. Nieprawidłowy obraz TKWR płuc występuje u ponad 85% chorych, a zmiany w badaniach czynnościowych układu oddechowego rozpoznawane są u 45-100% pacjentów, z czego u 25-41% chorych występują typowe zaburzenia restrykcyjne z obniżeniem FVC, TLC (całkowita pojemność płuc – total lung capacity) oraz upośledzeniem zdolności dyfuzyjnej płuc dla tlenku węgla (DLCO – diffusing lung capacity for carbon monoxide) (6, 12). U 18-47% chorych z chorobą śródmiąższową płuc zdarza się izolowane obniżenie zdolności dyfuzyjnej płuc (6) – należy pamiętać, że stwierdza się je również w izolowanym nadciśnieniu płucnym. Na podstawie rodzaju i lokalizacji zmian w obrazie tomokomputerowym można przewidzieć typ histopatologiczny zmian. W badaniu histopatologicznym wycinków płuc pacjentów chorych na twardzinę układową dominuje NSIP (nonspecific interstitial pneumonia – niespecyficzne śródmiąższowe zapalenie płuc) – obraz ten stwierdza się u 77,5% chorych, z czego typ komórkowy występuje u 24% pacjentów, a włókniejący w 76% przypadków, jedynie u 8% stwierdza się obraz UIP (ustal interstitial pneumonia – zwykłe śródmiąższowe zapalenie płuc) (13), sporadycznie w przebiegu sklerodermii dochodzi do rozwoju sarkoidozy (14), organizującego się zapalenia płuc (15) (OP – organising pneumonia) oraz krwawienia pęcherzykowego (16).
Obraz histologiczny zmian nie ma wpływu na rokowanie. 3 i 5-letnie przeżycie chorych z obrazem NSIP nie różni się istotnie od chorych z UIP, nie wykazano również istotnych statystycznie różnic w zakresie długości życia pomiędzy chorymi z komórkowym i włókniejącym typem NSIP. Niezależnymi niekorzystnymi czynnikami rokowniczym okazały się: eozynofilia powyżej 5% w płynie z płukania oskrzelowo-pęcherzykowego (BAL – bronchoalveolar lavage), niska wyjściowa wartość DLCO oraz spadek DLCO w ciągu pierwszych trzech latach obserwacji (13).
Zahamowanie zmian śródmiąższowych w płucach jest możliwe we wczesnej i aktywnej fazie choroby. Lekiem z wyboru, stosowanym od ok. 15 lat jest cyklofosfamid (1). W ostatnich latach przeprowadzono 2 wieloośrodkowe randomizowane, kontrolowane placebo badania dotyczące leczenia choroby śródmiąższowej płuc u chorych na twardzinę układową. W badaniu Scleroderma Lung Study oceniano efekt 12-miesięcznej terapii cyklofosfamidem doustnie vs placebo w grupie 158 chorych na sklerodermię z wczesną postacią choroby śródmiąższowej płuc. Odnotowano istotny statystycznie wzrost w zakresie FVC o 2,53% i TLC o 4,09% w grupie leczonej cyklofosfamidem w porównaniu do grupy przyjmującej placebo (17). W grupie leczonej obserwowano również zmniejszenie stwardnienia skóry, nasilenia duszności oraz poprawę jakości życia. W badaniu FAST (Fibrosing Alveolitis In Scleroderma Trial) badano wpływ leczenia glikokortykosteroidów i cyklofosfamidu podawanego drogą dożylną, które następnie kontynuowano azatiopryną vs placebo w grupie 45 chorych na twardzinę układową z chorobą śródmiąższową płuc. Po 12 miesiącach zaobserwowano tendencję do poprawy w zakresie FVC (wzrost o 2,4%) w grupie leczonej i spadek FVC o 3% w grupie placebo (18). W obydwu badaniach nie odnotowano różnic w zakresie zdolności dyfuzyjnej płuc. W innym retrospektywnym badaniu wykazano skuteczność azatiopryny i niskich dawek prednizolonu (19). Innym bezpiecznym, dobrze tolerowany lekiem immunosupresyjnym alternatywnym dla cyklofosfamidu pozostaje mykofenolan mofetilu (MMF). W grupie 9 chorych leczonych MMF i prednizonem w dawce 10 mg/d, zaobserwowano wzrost w zakresie FVC, TLC i DLCO (20). Wyniki badania są zachęcające, dlatego celowe i konieczne jest przeprowadzenie dużych randomizowanych, kontrolowanych placebo badań dotyczących skuteczności MMF. U pacjentów bez innych zmian narządowych zawsze należy też rozważyć wskazania do przeszczepienia płuc – wyniki dotychczas przeprowadzonych zabiegów są porównywalne do wyników transplantacji płuc przeprowadzanych z innych powodów (6, 21).
Zapalenie wielomięśniowe i skórnomięśniowe (PM/DM – polymyositis/dermatomyositis)
Zapalenia wielomięśniowe (PM) i skórnomięśniowe (DM) należą do miopatii zapalnych – układowych chorób tkanki łącznej o nieznanej etiologii charakteryzujących się osłabieniem siły mięśni proksymalnych oraz zajęciem różnych narządów wewnętrznych, w tym często płuc. Częstość występowania PM/DM wynosi 2-10/1 mln/rok (22), chorują przeważnie dzieci oraz dorośli w wieku 50-70 lat (23).
Najczęstszą płucną manifestacją PM/DM są zmiany śródmiąższowe. Na podstawie objawów klinicznych i zmian w klasycznym radiogramie klatki piersiowej oraz wyników badań czynnościowych można je stwierdzić u 5-46% chorych (4, 24, 25), na podstawie badania TKWR płuc zmiany śródmiąższowe w płucach są wykrywane nawet u ponad 70% chorych (26). Ich wystąpienie pogarsza przebieg choroby i rokowanie. Etiologia i patogeneza zmian śródmiąższowych w płucach nie są znane. Sugeruje się, że do ich rozwoju mogą przyczyniać się zakażenia (bakteryjne i wirusowe – wirus Epsteina-Baar, CMV, WZW typu C), leki, substancje chemiczne oraz mechanizmy immunologiczne i hormonalne. Bardziej predysponowane do rozwoju zmian śródmiąższowych są kobiety w wieku ok. 50 lat (23, 27). Istotnym czynnikiem predykcyjnym wystąpienia zapalenia śródmiąższowego płuc w PM/DM jest obecność przeciwciał anty Jo-1 (z grupy przeciwciał przeciwko syntetazie histydylowej tRNA).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Mouthon L, Berenze A, Guillevin L et al.: Therapeutic options for systemic sclerosis related interstitial lung diseases. Resp Med 2010; 104: 559-569.
2. Crestani B: The respiratory system in connective tissue disorders. Allergy 2005; 60: 715-734.
3. Diot E, Giraudeau B, Diot P et al.: Is anti-topoisomerase I a serum marker of pulmonary involvement in systemic sclerosis? Chest 1999; 116: 715-720.
4. Goh NS: Connective tissue disease and the lung. Clin Pulm Med 2009; 16: 309-314.
5. Strange CH, Highland KB: Interstitial lung disease in the patient who has connective tissue disease. Clin Chest Med 2004; 25: 549-559.
6. Highland KB, Garin MC, Brown KK: The spectrum of scleroderma lung disease. Semin Respir Crit Care Med 2007; 28: 418-429.
7. Steen VD, Medsger TA: Changes in causes of death in systemic sclerosis, 1972-2002. Ann Rheum Dis 2007; 66: 940-944.
8. Steen VD, Conte C, Owens GR et al.: Severe restrictive lung disease in systemic sclerosis. Arthritis Rheum 1994; 37: 1283-1289.
9. Gilson M, Zerkak D, Wipff J et al.: Prognostic factors for lung function in systemic sclerosis: prospective study of 105 cases. Eur Respir J 2010; 35: 112-117.
10. Savarino E, Bazzica M, Zentilin P et al.: Gastroesophageal reflux and pulmonary fibrosis in scleroderma. Am J Respir Crit Care Med 2009; 179: 408-413.
11. Goh NS, Desai SR, Veeraraghavan S et al.: Interstitial lung disease in systemic sclerosis: a simple staging system. Am J Respir Crit Care Med 2008; 177 (11): 1248-1254.
12. Ostojic P, Cerinic MM, Silver R et al.: Interstitial lung disease in systemic sclerosis. Lung 2007; 185: 211-220.
13. Bouros D, Wells AU, Nicholson AG et al.: Histopatologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med 2002; 165: 1581-1586.
14. Cox D, Conant E, earle L et al.: Sarcoidosis in systemic sclerosis: raport of 7 cases. J Rheumatol 1995; 22: 881-885.
15. Taylor JG, Bolster MB: Bronchiolitis obliterans with organizing pneumonia associated with scleroderma and scleroderma spectrum diseases. J Clin Rheumatol 2003; 9: 239-245.
16. Kallenbach J, Prinsloo I, Zwi S et al.: Progressive systemic sclerosis complicated by diffuse pulmonary haemorrhage. Thorax 1977; 32: 767-770.
17. Tashkin DP, Elashoff R, Clements PJ et al.: Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med 2006; 354: 2655-2666.
18. Hoyles RK, Ellis RW, Wellesbury J et al.: A multicenter, prospective, randomized, double-blind, placebo-controlledtrial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum 2006; 54: 3962-3970.
19. Dheda K, Lalloo UG, Cassim B et al.: Experience with azathioprine in systemic sclerosis associated with interstitial lung disease. Clin Rheumatol 2004; 23: 306-309.
20. Swigris JJ, Olson AL, Fischer A et al.: Mycophenolate mofetil is safe, well tolerated and preserves lung function in patients with connective tissue disease – related interstitial lung disease. Chest 2006; 130: 30-36.
21. Saggar R, Khanna D, Furst DE et al.: Systemic sclerosis and bilateral lung transplantation: a single centre experience. Eur Respir J 2010; 36: 893-900.
22. Kalluri M, Oddis ChV: Pulmonary manifestation of the idiopathic inflamatory myopathies. Clin Chest Med 2010; 31: 501-512.
23. Fathi M, Lundberg IE, Tornling G: Pulmonary complications of polymyositis and dermatomyositis. Semin Respir Crit Care Med 2007; 28: 451-458.
24. Douglas WW, Tazelaar HD, Hartman TE et al.: Polymyositis-dermatomyositis-associated interstitial lung disease. Am j Crit Care Med 2001; 164: 1182-1185.
25. Fathi M, Lundberg IE: Interstitial lung disease in polymyositis and dermatomyositis. Curr Opin Rheumatol 2005; 17: 701-706.
26. Olson AL, Brown KK: Connective tissue disease-associated lung disorders. Eur Respir Mon 2009; 46: 225-250.
27. Hirakata M, Nagai S: Interstitial lung disease in polymyositis and dermatomyositis. Curr Opin in Rheumatol 2000; 12: 501-508.
28. Selva-OCallaghan A, Labrador-Horillo M, Solans-Laque R et al.: Myositis-specific and myositis-associated antibodies in a series of eighty-eight Mediterranean patients with idiopathic inflammatory myopathy. Arthtitis Rheum 2006; 55: 791-798.
29. Bandoh S, Fujita J, Ohtsuki Y et al.: Sequential changes of KL-6 in sera of patients with interstitial pneumonia associated with polymyositis/dermatomyositis. Ann Rheum Dis 2000; 59: 257-262.
30. Fujita J, Dobashi N, Tokuda M et al.: Elevation of cytokeratin 19 fragment in patients with interstitial pneumonia associated with polymyositis/dermatomyositis. J Rheumatol 1999; 26: 2377-2382.
31. Mukae H, IshimotoH, Sakamoto N et al.: Clinical differences between interstitial lung disease associated with clinically amyopatic dermatomyositis and classic dermatomyositis. Chest 2009; 136: 1341-1347.
32. Cottin V, Thivolet-Bejui F, Reynaud-Gaubert M et al.: Interstitial lung disease in amyopatic dermatomyositis, dermatomyositis and polymyositis. Eur Respir J 2003; 22: 245-250.
33. Marie I, Hachulla E, Charin P et al.: Interstitial lung disease in polymyositis and dermatomyositis. Arthritis Rheum 2002; 47: 614-622.
34. Schnabel A, Reuter M, Biedrere J et al.: Interstitial lung disease in polymyositis and dermatomyositis: clinical course and response to treatment. Semin Arthritis Rheum 2003; 32: 273-284.
35. Tazelaar HD, Viggiano RW, Pickersgill J et al.: Interstitial lung disease in polymyositis and dernatomyositis: clinical features and prognosis as correlated with histologic findings. Am Rev Respir Dis 1990; 141: 727-733.
36. Pantin CFA: BTS statement on criteria for specialist referral, admission, discharge and follow-up for adults with respiratory disease. Thorax 2008; 63 (suppl 1): i1-i16.
37. Oddis CV, Sciurba FC, Elmagd KA et al.: Tacrolimus with refraktory polymyositis with interstitial lung disease. Lancet 1999; 353: 1762-1763.
38. Wikes M, Sereika S, Fertig N et al.: Treatment of anisyntethase associated interstitial lung disease with tacrolimus in patients with idiopathic inflammatory myopathy. Arthritis Rheum 2003; 48 (suppl): s434.
39. Antoniou KM, Margaritopoulos G, Economidou F et al.: Pivotal clinical dilemans in collagen vascular diseases associated with interstitial lung involvement. Eur Respir J 2009; 33: 882-896.
40. Papiris SA, Tsonis IA, Moutsopoulos HM et al.: Sjögrens syndrome. Semin Respir Crit Care Med 2007; 28: 459-472.
41. Kokosi M, Riemer EC, Highland KB: Pulmonary involvement in Sjögren Syndrome. Clin Chest Med 2010; 31: 489-500.
42. Parambil JG, Myers JL, Lindell RM et al.: Interstitial Lung Disease in Primary Sjögren syndrome. Chest 2006; 130: 1489-1495.
43. Papathanasiou MP, Constantopoulos SH, Tsampoulas C et al.: Reappraisal if respiratory abnormalities in primary and secondary Sjögrens syndrome. A controlled study. Chest 1986; 90: 370-374.
44. Cain HC, Noble PW, Matthay RA: Pulmonary manifestations of Sjögrens syndrome. Clin Chest Med 1988; 19: 687-699.
45. Tansey D, Wells AU, Colby TV et al.: Variations of histological patterns of interstitial pneumonia between connective tissue disorders and their relationship to prognosis. Histopatology 2004; 44: 585-596.
46. Ito I, Nagai S, Kitaichi M et al.: Pulmonary manifestations of primary Sjögrens syndrome: a clinical, radiologic and pathologic study. Am J Respir Crit Care Med 2005; 171: 632-638.
47. Devaraj A, Wells AU, Hansell DM: Computed tomographic imaging in connective tissue diseases. Semin Respir Crit Care Med 2007; 28: 389-397.
48. Kozuka T, Johkoh T, Honda O et al.: Pulmonary involvement in mixed connective tissue disease. J Thoracic Imaging 2001; 16: 94-98.
