© Borgis - Postępy Nauk Medycznych 4/2011, s. 333-336
Ewa Wolska1, *Ryszard Gellert1, Danuta Kobus1, Dorota Daniewska1, Andrzej Mróz2, Krzysztof Bardadin2, Agnieszka Perkowska-Ptasińska3
Torbielowatość kłębuszków nerkowych z towarzyszącym włóknieniem śródmiąższu nerek u pacjentki z nawrotową ich niewydolnością
Glomerulocystic kidney disease (GCKD) and tubulointerstitial fibrosis in a patient with recurrent acute kidney injure
1Department of Nephrology, Postgraduate Education Medical Center
Head of Department: prof. dr hab. med. Ryszard Gellert
2Department of Pathology, Postgraduate Education Medical Center
Head of Department: dr med. Krzysztof Bardadin
3Institute of Transplantology, Warsaw Medical University
Head of Institute: prof. dr hab. med. Magdalena Durlik
Streszczenie
Przedstawiamy przypadek 23-letniej kobiety, u której w biopsji nerki, wykonanej z powodu narastającej niewydolności nerek, uwidoczniono cechy torbielowatości kłębuszków nerkowych z towarzyszącym miernie rozległym włóknieniem zrębu i zanikiem cewek, co w koincydencji stanowi stosunkowo rzadkie, sporadycznie opisywane w literaturze znalezisko.
Wszelkie próby jakiegokolwiek postępowania farmakologicznego nieuchronnie prowadziły do rozwoju ostrego uszkodzenia nerek (AKIN I-II). Sądzimy, iż podobne epizody AKI mogły mieć miejsce w przeszłości, co w konsekwencji doprowadziło do śródmiąższowego włóknienia nerek.
Nieproporcjonalnie wysokie stężenia kwasu moczowego mogło pośrednio wskazywać na zaburzenie syntezy/sekrecji uromoduliny typowe dla innych ciliopatii.
Obraz histopatologiczny w powiązaniu z przebiegiem klinicznym GCKD u opisywanej pacjentki może sugerować bezpośredni związek pomiędzy ciliopatiami a zwiększoną podatnością na występowanie AKI, które w konsekwencji może prowadzić do włóknienia śródmiąższowego i najprawdopodbniej przyspiesza rozwój niewydolności nerek.
Summary
We present a case of glomerular cystic kidney disease, most probably sporadic, revealed on renal biopsy performed to diagnose the deteriorating renal function in a 23 years old female. Surprisingly, the GCKD was accompanied by mild tubolointerstitial fibrosis, which is unusual and has been reported sporadically. Any farmacological intervention inevitably led to acute kidney injury (AKIN I-II). We hypothesize, that this could have happened also prior to diagnosis and the previous recurrent AKI episodes resulted in interstitial fibrosis. The inappropriately increased plasma uric acid concentrations could indicate the disturbance in uromodulin synthesis/secretion, which is typical to other ciliopathies. The unusual findings on renal biopsy and clinical course of GCKD in patient suggests direct link between ciliopathies and susceptibility to acute kidney injury, which can result in interstitial fibrosis and possibly, accelerated kidney failure progression.

Introduction
Glomerulocystic Kidney disease (GCKD) is a relatively uncommon type of renal cystic disease. GCKD is more frequently diagnosed in children, however, it can be dignosed at any age. It is believed that the prevalence of GCKD is underestimated among adult patients presenting with end – stage renal failure.
We report a case of GCKD first diagnosed in the age of 23-year-in a female with chronic kidney disease (stage III).
Case report
A 23-year-old female with a 16-year history of normocytic anemia was adimtted for evaluation of an accidentally diagnosed renal insufficiency. The patient denied an infection, fever or drug abuse and difficulty or pain in urination preceding the hospitalization. Prior to the admission her only medication had been oral contraceptive. She reported past recurrent urinary tract infections with no history of haematuria or lithuria. She also had a bone marrow biopsy in 1994 and oral iron therapy, but the documentary evidence was no longer available.
There was no family history of renal insufficiency, however, her father suffered from nephrolithiasis.
On admission the patient was in good general condition without any complaints. She showed a marked pallor of the facial skin and mucous membranes. The remainder of the physical examination was normal. Her blood pressure was 110/80 mm and the heart rate 72 beats/min.
Laboratory tests indicated an impaired renal function with a serum creatinine of 1.53 mg/dl, serum urea 52.6 mg% and GFR 43 ml/min, serum sodium 138 mmol/l, serum potasium 5.6 mmol/l. Moreover, the hemoglobin level was moderately decreased – 10.0 g/L, MCV, MCH, MCHC, the reticulocyte count (0.7%), the total leucocyte count and platelet count were within normal limit, The TSAT was 28%, and the concentrations of Vit B12 and Folic Acid, Serum phosphorus (3.66 mg/dl) and fasting plasma glucose (99 mg/dl) were normal. Venous blood gases indicated a metabolic acidosis with pH 7.334, pCO2 31.7 mmHg, pO2 26.8 mmHg, HCO3 – 16,9 mmol/l, BE-7,5 mmol/l with normal serum anion gap. Howerer, the significant increase in uric acid – 6.78 mg /dl, and dyslipidemia: total cholesterol 245 mg/dl, LDL-cholesterol – 135 mg/dl, HDL – cholesterol 78.9 mg/dl, Triglycerides 80.72 mg/dl and ESR of 50 mm were noted.
Routine immunological screening (c-ANCA, p-ANCA, ANA, DsDNA, anty GBM, RF) was negative. Thyrothropin as well as thyroid hormones were normal.
Urinalysis was normal with pH 5.0 and specific gravity 1015 without proteinuria or erythrocyturia.
Ultrasound examination revealed a slightly diminished kidneys measuring – right kidney 9.4 cm and the left 9.2 cm, with mild cortical hyperechogenicity and the only one cortical cyst of 1.3 cm in the left kidney. No signs of obstruction and no cysts in any other organs were found.
CT scanning was not performed due to renal insufficiency.
A percutaneous renal biopsy was done to show six glomeruli with cystic dilatation of Bowman’s space, compression of the capillary loops and mild tubulointerstitial fibrosis. Immunofluorescence studies were negative. The histopatologic findings were consistent with GCKD with concomitant tubulointerstitial fibrosis (fig. 1).
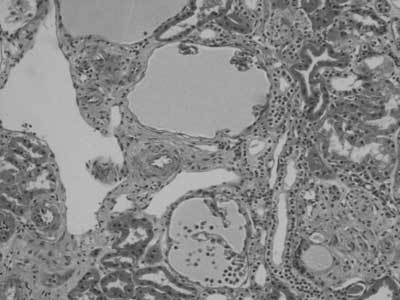
Fig. 1.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Bernstein J: Glomerulocystic kidney disease – nosological considerations. Pediatr Nephrol 1993; 7: 464-470.
2. Sharp CK, Bergman SM, Stockwin JM et al.: Dominantly transmitted glomerulocystic kidney disease: a distinct genetic entity. J Am Soc Nephrol 1997; 8: 77-84.
3. Bissler J, Siroky B, Yin H: Glomerulocystic kidney disease.Educational review. Pediatr Nephrol 2010; 25: 2049-2059.
4. Gusmano R, Caridi G, Marini M et al.: Glomerulocystic kidney disease in a family. Nephrol Dial Transplan 2002; 17: 813-818.
5. Abderrahim E, Moussa F, AbdallahTB et al.: Glomerulocystic kidney disease in an adukt presenting as and -stage renal failure. Nephrol Dial Transplant 1999; 14: 1276-1278.
6. Meral Gunay-Aygun: Liver and kidney disease in ciliopathies. Am J Med Genet C Semin Med Genet 2009; 15: 269-306.
7. Zaucke F, Boenhlein JM, Steffens S et al.: Uromodulin is expressed in renal primary cilia and UMOD mutations result in decreased ciliary uromodulin expression. Hum MOL Genet 2010; 19: 1985-97.
8. Izzi c, Sottini L, Dallera N, Capistrano M et al.: Genetics and nosological classification of renal cystic diseases. G Ital Nefrol 2010; 27 (Suppl 50): 63-9.
9. Flavio Teles de Farias Filho, Americo Cuvello Neto, Regina CRM: Abdulkader. Glomerulocystic kidney disease presenting as acute renal failure in an adult patient. Nephrol Dial Transplant 2005; 20: 2293.
10. Thompson SJ, Morley AR: Glomerulocystic Kidney Disease Associated with Haemolytic-Uraemic Syndrome. Nephrol Dial Transplant 1991; 6: 131-133.
11. Gersh M, Mutig K, Bachmann s et al.: Is salt-wasting the long awaited answer to the hyperuricaemia seen in uromodulin storage diseases? Nephrol Dial Transplant 2006; 21: 2028-29.
12. Bingham C, Bulman M, Ellard S et al.: Mutations in the hepatocyte nuclear factor – 1 beta gene are associated with familial hypoplastic glomerulocystic kidney disease. Am J Hum Genet 2001; 68: 219-224.
13. Baxter T: Cysts arising in the renal corpuscle Arch Dis Child 1965; 40: 455-463.
14. Kirti Gupta, Mahesha Vankalakunti, Man Updesh Singh Sachdeva: Glomerulocystic Kidney Disease and its rare associations: an autopsy report of two unrelated cases. Diagn Pathol 2007; 2-12.
15. Jain M, LeQuesne GW, Bourne AJ, Henning P: High-resolution ultrasonography in the differential diagnosis of cystic diseases of the kidney in infancy and childhood: preliminary experience. J Ultrasound Med 1997; 16: 235-240.
16. Crowe AV, Woolfson RG, Griffiths MH, Neild GH: Glomeruloystic kidney disease associated with Wegener’s granulometosis and membranous glomerulonephritis: a case report. Nephrol Dial Transplant 1995; 10: 888-890.
17. Sahay M, Swarnalata G: Gromelurocystic kidney disease. NDT Plus 2010; 3: 349-350.
