© Borgis - Postępy Nauk Medycznych 6/2011, s. 512-520
*Grzegorz Helbig, Sławomira Kyrcz-Krzemień
Aktualne postępy w patogenezie i leczeniu klasycznych nowotworów mieloproliferacyjnych Filadelfia-ujemnych
Recent advances in pathogenesis and treatment of classical Philadelphia-negative myeloproliferative neoplasms
Department of Hematology and Bone Marrow Transplantation, Silesian Medical University, Katowice
Head of Department: prof. Sławomira Kyrcz-Krzemień
Streszczenie
W ostatnich kilku latach jesteśmy świadkami ogromnego postępu jaki dokonał się w zrozumieniu patogenezy nowotworów mieloproliferacyjnych (MPNs). Klasyczne nowotwory mieloproliferacyjne Filadelfia-ujemne obejmują nadkrwistość prawdziwą, nadpłytkowość samoistną i pierwotne włóknienie szpiku. Wszystkie te jednostki chorobowe wykazują podobieństwo obrazu klinicznego i zostały zgrupowane razem przez Damesheka w roku 1951. Od tego czasu, w związku z odkryciem mutacji JAK2V617F i MPL zmieniło się nasze spojrzenie na patogenezę tych chorób i doprowadziło do rewizji dotychczasowych kryteriów diagnostycznych Światowej Organizacji Zdrowia (WHO). Nowa klasyfikacja WHO została zaproponowana w roku 2008 i objęła ona niedawno opisane nieprawidłowości molekularne, zmieniając zasadniczo aktualne podejście diagnostyczne. Co więcej, mutacje te zmodyfikowały sposób monitorowania chorób, oceny efektów leczenia, a także stały się potencjalnym terapeutycznym celem dla nowych cząsteczek. W pracy przedstawiono aktualne spojrzenie na molekularną patogenezę MPNs, z podkreśleniem znaczenia nowych mutacji genetycznych. Poddano dyskusji kryteria diagnostyczne i dostępne opcje lecznicze.
Summary
In the last few years we have witnessed a great progress in our understanding of the pathogenesis of myeloproliferative neoplasms (MPNs). The classical Philadelphia-negative MPNs include polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF). They were grouped together based on similar clinical phenotype by Dameshek in 1951. Since then the identification of JAK2V617F and MPL mutations has changed our view on disease pathogenesis and led to the revision of World Health Organization (WHO) diagnostic criteria for classical Philadelphia-negative MPNs. A new classification issued in 2008 incorporated these molecular abnormalities and therefore has modified the diagnostic approach. Moreover, these mutations have modified the strategies for monitoring and response assessment and they could become the potential targets for small-molecule agents. In this review we present current overview on molecular basis of MPNs including novel mutations and discuss the diagnostic criteria and therapeutic modalities.

Introduction
The myeloproliferative neoplasms (MPNs) comprise a group of clonal hematopoietic stem cell disorders characterized by proliferation of one or more myeloid lineage. MPNs occur mainly in adults between 5th and 7th decade of life, and the annual incidence is estimated to be 6-10 per 100 000 population (1). The term “myeloproliferative disorders” (MPDs) was proposed in 1951 by Dameshek to encompass several disorders with similar clinical phenotype. They included polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF), chronic myelogenous leukemia (CML) and erythroleukemia (2). The last 5 years have brought a pivotal progress in our understanding of the pathogenesis of classical Ph-negative MPDs. It was associated with the identification of the specific molecular marker- the JAK2V617F point mutation, which seems to be involved in PV, ET and PMF (3). It is now clear that patients with this mutation are biologically distinct from those without the mutation and that the mutation is associated with different disease phenotype. Moreover, several novel mutations have been recently identified, but their pathogenic and prognostic relevance has not been established yet (4). The current World Health Organization (WHO) classification for hematological malignancies has incorporated the recently discovered mutations into the diagnostic approach. The term “MPDs” has been changed to “MPNs”. The WHO classification includes 8 clinical entities and it was presented in table 1 (5).
Table 1. The World Health Organization Classification of Myeloid Malignancies (5).
| Myeloproliferative neoplasms (MPNs) |
| I Classical MPNs |
Chronic myelogenous leukemia, BCR-ABL1 positive
Polycythemia veravEssential thrombocythemia
Primary myelofibrosis |
| II Non-classical MPNs |
Chronic neutrophilic leukemia
Chronic eosinophilic leukemia, not otherwise specified
Mastocytosis
Myeloproliferative neoplasms, unclassifiable |
This current review will focus on classical Ph-negative MPNs and includes molecular basis of pathogenesis, risk stratification, diagnostic and therapeutic management.
Molecular basis for myeloproliferative neoplasms
JAK2- mutated MPNs
JAK2 is located on chromosome 9p24 and it belongs to the Janus family of non-receptor protein tyrosine kinase. Janus kinase/signal transducer and activator of transcription (JAK-STAT) signaling remains crucial for survival and normal function of hematopoietic and other cells (6). The JAK2V617F mutation is of particular relevance to MPNs including mainly PV, ET and PMF (3). The JAK2V617F results from a somatic G to T mutation involving JAK2 exon 14. It leads to nucleotide shift at position 1849 and the substitution of valine to phenyloalanine at codon 617. Mutated JAK2 causes cytokine-independent growth of cells and their hypersensitivity to cytokines (7). The JAK2V617F point mutation is found in approximately 95% of patients with PV and 40-50% of patients with ET and PMF (3, 8). Two-step model may explain the development of cells which are homozygous for JAK2 mutation. Initially, the occurrence of the JAK2V617F gives rise to a heterozygous clone which replaces normal hematopoietic cells. The second step includes mitotic recombination within cells heterozygous for the JAK2V617F with subsequent uniparental disomy. As a consequence, the daughter cells are homozygous for JAK2V617F and they replace the heterozygous clone. It may happen that myeloid cell population includes a variable proportion of JAK2 mutant alleles; it means that cells with JAK2 mutation may coexist with cells lacking this abnormality. The first step is characterized by progressive increase of mutant alleles from 0 to 50%, whereas the second step from 50 to 100% (9). The experimental studies in mouse model demonstrated that both heterozygous and homozygous JAK2 mutations induce PV-like disease phenotype, but homozygous JAK2 mutation is associated with more aggressive disease with progression to post-PV myelofibrosis (10). The JAK2V617F homozygosity is frequently seen in PV and PMF. In contrast, high mutant allele burden is rarely seen in ET (11). Homozygosity in granulocytes can be detected in 30% of patients with PV (3), but it was demonstrated in approximately 90% of colonies of hematopoietic progenitor cells derived from patients with PV whereas homozygous progenitor colonies were not observed in ET (12). This difference in the occurrence of homozygosity may result from the substantial variation in the rate of mitotic recombination among persons and the role of genetic and environmental factors determining susceptibility to mitotic recombination is very likely (11). However, it should be clearly underlined that variability in the allele burden is not sufficient to distinguish different clinical entities. It does not help us to answer the question how one mutation gives rise to three different clinical phenotypes. Moreover, there is an increasing number of evidence that the JAK2V617F is not an initiating event in MPNs and that pre-JAK2 mutated clone may exist (13). Some studies suggest that deletions of 20q and other cytogenetic abnormalities are present in about 10% of patients with MPNs and that this deletion may occur before the JAK2 mutation. It was demonstrated that deletions 20q are exclusively V617F-positive (14). In conclusion, the pathogenic role of JAK2V617F mutation requires to be better defined, especially in the light of the presence of novel mutations which may occur in patients with MPNs (4).
As it was aforementioned, JAK2 allele burden is associated with clinical phenotype and PV patients homozygous for V617F mutation have been found to have a more severe disease (15). Recently published report has demonstrated that patients with an allele burden equal or > 50% had higher white blood cell (WBC) count, greater spleen size and lower platelet count than those with less than 50% of mutant dosage. It was also proved that patients homozygous for JAK2 mutation had significantly higher risk of developing myelofibrosis (16).
JAK2 exon 12 mutations were identified in PV who were negative for JAK2V617F point mutation. Those patients presented with an isolated erythrocytosis, distinct bone marrow morphology and low serum erythropoietin level. It is noteworthy that erythroid colonies have grown from their blood samples in the absence of exogenous erythropoietin. In contrast to JAK2V617F-positive PV patients, JAK2 exon 12 mutated PV subset is often heterozygous for this mutation (17).
MPL mutations
MPL (myeloproliferative leukemia virus oncogene) located on chromosome 1p34, is the thrombopoietin receptor. MPL is highly expressed in early hematopoietic progenitors and in cells of megakaryocytic lineage. MPL is the key growth and survival factor for megakaryocytes (18). The two most common MPL mutations are W515L and W515K. They were found in about 10% of patients with PMF and in less than 10% of ET-patients lacking the JAK2V617F mutation (19). MPL-mutated ET patients are older, have lower hemoglobin concentration, higher platelet count and higher risk of arterial thrombosis (20) whereas the presence of MPL mutation in patients with PMF is associated with female gender, older age, lower hemoglobin concentration and blood transfusion dependence (21).
TET2 mutations
TET2 is located on chromosome 4q24 and its function is probably associated with epigenetic regulation of transcription (22). TET2 mutations are present in about 10-15% of patients with MPNs, including both the JAK2V617F – positive and negative cases (23). It should be highlighted that TET2 mutations may coexist with other relevant mutation e.g. KITD816V for mast cell disease (24). Moreover, it was demonstrated that TET2 mutations may precede the acquisition of a JAK2 mutation. Up-to-date studies show that the presence of TET2 mutations in MPNs do not affect survival, leukemic transformation and the risk of thrombosis (23).
Novel mutations in MPNs
Several novel mutations have been recently found in patients with MPNs:
1) ASXL1 (Additional Sex Combs-Like 1,
2) CBL (Casitas B-lineage lymphoma proto-oncogene),
3) IDH1 and IDH2 (Isocitrate dehydrogenase 1 and 2),
4) IKZF1 (IKAROS family zinc finger 1),
5) LNK,
6) EZH2.
All these mutations are rarely seen in patients with chronic phase of MPNs whereas their frequency may exceed 20% in blast transformation (tab. 2) (4).
Table 2. Novel mutations in classical myeloproliferative neoplasms (4,43) modified by authors.
| Mutation | Chromosome location | Mutational frequency |
JAK2 (Janus kinase 2)
JAK2V617F exon 14 | 9p24 | PV~96%; ET~55%; PMF~65%; BP~50% |
| JAK2 exon 12 | 9p24 | PV~3% |
| MPL (myeloproliferative leukemia virus oncogene) exon 10 | 1p34 | ET~3%, PMF~10%; BP~5% |
| TET2 (TET oncogene family member 2) exon 12 | 4q24 | PV~16%; ET~5%; PMF~17%; BP~17% |
| ASXL1 (additional sex combs-like-1) exon 12 | 20q11.1 | PMF~13%, BP~18% |
| CBL (casitas B-lineage lymphoma proto-oncogene) exon 8/9 | 11q23.3 | PMF~6% |
| IDH1/IDH2 (isocitrate dehydrogenease) exon 4 | 2q33.3/15q26.1 | PV~2%, ET~1%, PMF~4%, BP~20% |
| IKZF1 (IKAROS family zinc finger 1) | 7p21 | BP~19% |
| LNK exon 2 | 12q24.12 | BP~10% |
| EZH2 (enhancer of zeste homolog 2) exons 10, 18 and 20 | 7q36.1 | PV~3%, PMF~7% |
Legend: PV = polycythemia vera, ET = essential thrombocythemia, PMF = primary myelofibrosis, BP = blast phase.
The results including the analysis of JAK2 haplotype may suggest that hereditary component confers susceptibility to MPNs and some of above-mentioned mutations may play a role at different stages of disease evolution. A strong association between the risk of developing a JAK2V617F-positive MPNs and a germline haplotype (46/1 or GGCC) has been recently demonstrated (fig. 1) (25).
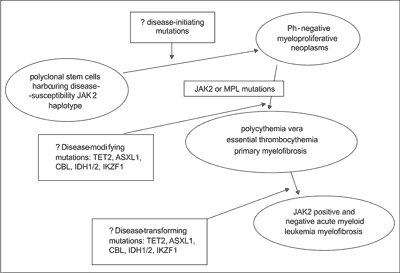
Fig. 1. Clonal evolution in myeloproliferative neoplasms (modified from 4).
2008 WHO diagnostic criteria for MPNs
General aspects
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Vardiman JW, Brunning RD, Arber DA et al.: Introduction and overview of the classification of the myeloid neoplasms. [In:] Swerdlow SH, Campo E, Lee Harris N, et al. eds. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press 2008; 31-73.
2. Dameshek W: Some speculations on the myeloproliferative syndromes. Blood 1951; 6: 372-375.
3. Baxter EJ, Scott LM, Campbell PJ et al.: Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365: 1054-1061.
4. Tefferi A: Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZF1. Leukaemia 2010; 24: 1128-1138.
5. Swerdlow SH, Campo E, Harris NL et al.: WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press 2008.
6. Murray PJ: The JAK-STAT signaling pathway: input and output integration. J Immunol 2007; 178: 2623-2629.
7. Kilpivaara O, Levine RL: JAK2 and MPL mutations in myeloproliferative neoplasms: discovery and science. Leukemia 2008; 22: 1813-1817.
8. Jones AV, Kreil S, Zoi K et al.: Widespread occurrence of the JAK2V617F mutation in chronic myeloproliferative disorders. Blood 2005; 106: 2162-2168.
9. Kralovics R, Passamonti F, Buser AS et al.: A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med 2005; 352: 1779-1790.
10. Vanunucchi AM, Antonioli E, Guglielmelli P et al.: Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia 2007; 21: 1951-1959.
11. Campbell PJ, Green AR: The myeloproliferative disorders. N Engl J Med 2006; 355: 2451-2466.
12. Scott LM, Scott MA, Campbell PJ et al.: Progenitors homozygous for the V617F JAK2 mutation occur in most patients with polycythemia vera but not essential thrombocythemia. Blood 2006; 108: 2435-2437.
13. Skoda R: The genetic basis of myeloproliferative disorders. Hematology Am Soc Hematol Educ Program 2007; 2007: 1-10.
14. Campbell PJ, Baxter EJ, Beer PA et al.: Mutation of JAK2 in the myeloproliferative disorders: timing, clonality studies, cytogenetic associations and role in leukemic transformation. Blond 2006; 108: 3548-3555.
15. Vannucchi AM, Antonioli E, Guglielmelli P et al.: Clinical profile of homozygous JAK2 617V > F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007; 110: 840-846.
16. Passamonti F, Rumi E, Pietra D et al.: A prospective study of 338 patients with polycythemia vera: the impact of JAK2(V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia 2010; 24: 1574-1579.
17. Scott LM, Tong W, Levine RL et al.: JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007; 356: 459-468.
18. Kaushansky K: The molecular mechanisms that control haematopoiesis. J Clin Invest 2005; 115: 3339-3347.
19. Vannucchi AM, Guglielmelli P, Tefferi A: Advances in understanding and management of myeloproliferative neoplasms. Ca Cancer J Clin 2009; 59: 171-191.
20. Beer PA, Campbell PJ, Scott LM et al.: MPL mutations in myeloproliferative disorders: analysis of the PT-1 cohort. Blood 2008; 112: 141-149.
21. Guglielmelli P, Pancrazzi A, Bergamaschi G et al.: Anaemia characterizes patients with myelofibrosis harbouring Mpl mutation. Br J Haematol 2007; 137: 244-247.
22. Tahiliani M, Koh KP, Shen Y et al.: Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by TET1. Science 2009; 324: 930-935.
23. Delhommeau F, Dupont S, Della Valle V et al.: Mutation in TET2 in myeloid cancers. N Engl J Med 2009; 360: 2289-2301.
24. Tefferi A, Levine RL, Lim KH et al.: Frequent TET2 mutations in systemic mastocytosis: clinical, KITD816F and FIP1L1-PDGFRA correlates. Leukemia 2009; 23: 900-904.
25. Olcaydu D, Harutyunyan A, Jager R et al.: A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat Genet 2009; 41: 450-454.
26. Passamonti F, Rumi E, Pietra D et al.: JAK2 (V617F) mutation in healthy individuals. Br J Haematol 2007; 136: 678-679.
27. Sidon P, El Housni H, Dessars B et al.: The JAK2V617F mutation is detectable at very low level in peripheral blood of healthy donors. Leukemia 2006; 20: 1622.
28. Wadleigh M, Tefferi A: Classification and diagnosis of myeloproliferative neoplasms according to the 2008 World Health Organization criteria. Int J Hematol 2010; 91: 174-179.
29. Marchioli R, Finazzi G, Landolfi R et al.: Vascular and neoplastic risk in a large cohort of patients with polycythemia vera. J Clin Oncol 2005; 23: 2224-2232.
30. McMullin MF, Bareford D, Campbell P et al.: Guidelines for the diagnosis, investigation and management of polycythemia/erythrocytosis. Br J Haematol 2005; 130: 174-195.
31. Finazzi G, Barbui T. How I treat patients with polycythemia vera. Blood 2007; 109: 5104-5111.
32. Cervantes F, Passamonti F, Barosi G et al.: Life expectancy and prognostic factors in the classic BCR/ABL-negative myeloproliferative disorders. Leukemia 2008; 22: 905-914.
33. Jensen MK, de Nully BP, Nielsen OJ et al.: Incidence, clinical features and outcome of essential thrombocythemia in a well defined geographical area. Eur J Haematol 2000; 65: 132-139.
34. Harrison CN, Campbell PJ, Buck G et al.: Hydroxyurea compared to anagrelide in high risk essential thrombocythemia. N Engl J Med 2005; 353: 33-45.
35. Gisslinger H: Update on diagnosis and management of essential thrombocythemia. Semin Thromb Hemost 2006; 32: 430-436.
36. Carobbio A, Antonioli E, Guglielmelli P et al.: Leukocytosis and risk stratification assessment in essential thrombocythemia. J Clin Oncol 2008; 26: 2732-2736.
37. Harrison C: Management of essential thrombocythemia: lessons from the Primary-Thrombocythemia 1 trial. Haematologica 2010; 4: 197-203.
38. Budde U, Schaefer G, Mueller N et al.: Acquired von Willebrand’s disease in the myeloproliferative syndrome. Blood 1984: 64: 981-985.
39. Tefferi A: Myelofibrosis with myeloid metaplasia. N Engl J Med 2000; 342: 1255-1265.
40. Dupriez B, Morel P, Demory JL et al.: Prognostic factors in agnogenic myeloid metaplasia: a report on 195 cases with a new scoring system. Blood 1996; 88: 1013-1018.
41. Cervantes F, Dupriez B, Pereira A et al.: New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009; 113: 2895-2901.
42. Gangat N, Caramazza D, Vaidya R et al.: DIPSS-Plus: a Refined Dynamic International Prognostic Scoring System (DIPSS) for Primary Myelofibrosis that incorporates prognostic information from karyotype, platelet count and transfusion status. J Clin Oncol 2010. Doi:10.1200/JCO.2010.32.2446.
43. Tefferi A: How I treat myelofibrosis. Blood 2011; Doi: 10.1182/blood-2010-11-315614.
44. Kroger N, Mesa RA: Choosing between stem cell therapy and drugs in myelofibrosis. Leukemia 2008; 22: 474-486.
45. Huang J, Tefferi A: Erythropoiesis stimulating agents have limited therapeutic activity in transfusion-dependent patients with primary myelofibrosis regardless of serum erythropoietin level. Eur J Haematol 2009; 83: 154-155.
46. Tefferi A, Lasho TL, Mesa RA et al.: Lenalidomide therapy in del(5)(q31)-associated myelofibrosis: cytogenetic and JAK2V617F molecular remissions. Leukemia. 2007; 21: 1827-1828.
47. Pardanani A, George G, Lasho T et al.: A Phase I/II Study of CYT 387, an oral JAK1/2 inhibitor, in myelofibrosis: significant response rates in anemia, splenomegaly and constitutional symptoms. ASH Annual Meeting Abstracts. November 20, 2010; 116: 460.
48. Tefferi A: Primary myelofibrosis. Cancer Treat Res 2008; 142: 29-49.
