© Borgis - Nowa Pediatria 4/2014, s. 128-131
Tomasz Książczyk, *Agnieszka Tomik, Małgorzata Gołąbek-Dylewska
Zwężenie cieśni aorty i dwupłatkowa zastawka aortalna u dziewczynki z zespołem Turnera – opis przypadku
Coarctation of the aorta and bicuspid aortic valve in a girl with Turner syndrome – a case report
Klinika Kardiologii Wieku Dziecięcego i Pediatrii Ogólnej, Warszawski Uniwersytet Medyczny
Kierownik Kliniki: prof. dr hab. n. med. Bożena Werner
Summary
The authors present a case of a 4-year-old girl with Turner syndrome (kariotype 45XO). The diagnosis was established prenatally in amniopuncture. Characteristic dysmorphic features coexisting hands and feet lymphatic oedema have been already seen in a newborn period. Echocardiography revealed coarctation of the aorta and bicuspid aortic valve. The patient's condition remains stable without arterial hypertension during follow-up. She is also under endocrinological care due to short stature. Diseases of the cardiovascular system are a crucial factor affecting life expectancy of patients with Turner syndrome. The most common, affecting about 45% of patients are bicuspid aortic valve and coarctation of the aorta as in the described case. There is a risk of dilation and dissecting of the aorta because of the congenital heart defects and high prevalence of arterial hypertension. For that reason girls with Turner syndrome should undergo detailed cardiovascular system evaluation including echocardiography and MRI, and be assessed by pediatric cardiologist on a regular basis.

WSTĘP
Zespół Turnera (TS) jest spowodowany całkowitym lub częściowym brakiem jednego z chromosomów X. Występuje u około 1/2000 żywo urodzonych dziewczynek, ale ryzyko urodzenia dziecka z TS nie rośnie wraz z wiekiem matki. Typowe dla tego zespołu są: wrodzona dysgenezja gonad, niedobór hormonów płciowych, niski wzrost, charakterystyczne cechy fenotypowe oraz wrodzone wady układu sercowo-naczyniowego, które w największym stopniu wpływają na długość życia pacjentek z TS.
OPIS PRZYPADKU
Dziewczynka z prenatalnym rozpoznaniem zespołu Turnera została przyjęta w 4. tygodniu życia do Kliniki Kardiologii ze szpitala powiatowego z powodu podejrzenia zwężenia cieśni aorty. Dziecko urodzone z ciąży I, porodu I, u którego w I trymestrze ciąży wysunięto podejrzenie wady genetycznej ze względu na obecność obrzęku karku w badaniu USG. W 17. tygodniu ciąży wykonano amniopunkcję, której wynik potwierdził obecność nieprawidłowego kariotypu: 45,X. Dziewczynka urodziła się w 40. tygodniu ciąży, z masą ciała 2670 g. Po urodzeniu obserwowano typowe cechy fenotypowe zespołu Turnera: nadmiar skóry na karku, skośne (antymongoidalne) ustawienie powiek, zmarszczki nakątne oraz duże obrzęki limfatyczne dłoni i stóp.
W 4. tygodniu życia dziecka rodzice zgłosili się z córką do szpitala z powodu objawów infekcji dróg oddechowych, gdzie rozpoznano zapalenie płuc i wdrożono antybiotykoterapię. Ze względu na obecność szmeru nad sercem wykonano badanie echokardiograficzne i wysunięto podejrzenie zwężenia cieśni aorty.
Przy przyjęciu dziewczynka była w stanie ogólnym dobrym, bez objawów niewydolności serca, w badaniu przedmiotowym zwracały uwagę znaczne obrzęki limfatyczne dłoni i stóp, nadmiar skóry na karku oraz dysmorfia twarzy. Czynność serca wynosiła 120/min, nad podstawą serca słyszalny był szmer skurczowy 2/6, promieniujący do okolicy międzyłopatkowej, tętno na tętnicach udowych obecne, ale wyraźnie słabsze niż na tętnicy ramieniowej. W pomiarach manometrycznych ciśnienia tętniczego krwi na kończynach górnych stwierdzono RR do 90/42 mm Hg, na kończynach dolnych 77/43 mm Hg, gradient ciśnienia skurczowego pomiędzy kończyną górną i dolną wynosił maksymalnie 13 mm Hg. Saturacja przezskórna wynosiła 98%. W ścisłym bilansie płynów stwierdzono właściwe dla wieku objętości przyjmowanego pokarmu oraz prawidłową diurezę.
W zapisie elektrokardiograficznym (EKG) stwierdzono rytm zatokowy o częstości 130/min, bez cech przerostu jam serca. W RTG klatki piersiowej wielkość serca oraz rysunek naczyniowy płuc były prawidłowe. Badanie echokardiograficzne wykazało: dwupłatkową zastawkę aorty czynnościowo prawidłową (ryc. 1), lewostronny łuk aorty ze zwężeniem w miejscu cieśni o ok. 30% w stosunku do wymiaru łuku aorty i przyspieszeniem przepływu, z maksymalnym gradientem skurczowym 30 mm Hg (ryc. 2A i B), pulsacyjny przepływ w aorcie brzusznej. Wielkość i kurczliwość lewej komory były prawidłowe.

Ryc. 1. ECHO-2D. Dwupłatkowa zastawka aortalna (BAV). LA – lewy przedsionek, RA – prawy przedsionek, RV – prawa komora, PA – tętnica płucna.
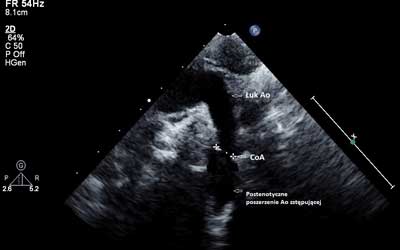
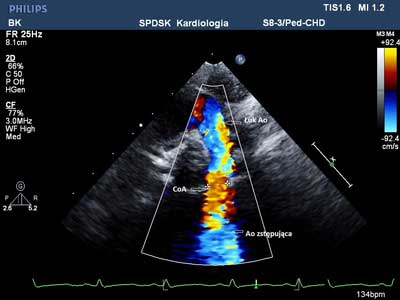
Ryc. 2. A – ECHO-2D. Zwężenie cieśni aorty (CoA) z postenotycznym poszerzeniem aorty zstępującej. B – turbulentny, przyspieszony przepływ przez cieśń aorty zarejestrowany techniką kolorowego Dopplera.
W badaniu ultrasonograficznym (USG) jamy brzusznej nie stwierdzono nieprawidłowości.
Na podstawie stanu klinicznego i wykonanych badań nie stwierdzono wskazań do interwencji kardiochirurgicznej.
Dziewczynka (obecnie 4,5-letnia) pozostaje pod opieką Kliniki Kardiologii, w kolejnych badaniach nie obserwowano narastania zwężenia w miejscu cieśni aorty ani narastania gradientu skurczowego przez miejsce zwężenia. Funkcja dwupłatkowej zastawki aortalnej jest prawidłowa. Cieśnienie tętnicze w pomiarze manometrycznym pozostaje < 90 percentyla dla płci i wieku, nie stwierdza się istotnej różnicy ciśnień pomiędzy kończynami górnymi i dolnymi. Aktualny wzrost to 97 cm, czyli < 3 percentyla dla płci i wieku. Rozwój psychoruchowy jest prawidłowy.
Ze względu na aberrację chromosomalną dziewczynka wymaga stałej wielospecjalistycznej opieki, w tym genetycznej i endokrynologicznej.
DYSKUSJA
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Kansra AR, Donohoue PA: Hypergonadotropic hypogonadism in the female. [In:] Kliegman RM, Stanton BF, Schor NF et al. (eds.): Nelson Textbook of Pediatrics. 19th ed., Elsevier, Philadelphia 2011: 1951-1954. 2. Gøtzsche CO, Krag-Olsen B, Nielsen J et al.: Prevalence of cardiovascular malformations and association with karyotypes in Turner’s syndrome. Arch Dis Child 1994; 71: 433-436. 3. Bondy CA: Turner Syndrome Study Group: Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group. Clin Endocrinol Metab 2007; 92: 10-25. 4. Carvalho AB, Guerra G, Baptista MT et al.: Cardiovascular and renal anomalies in Turner syndrome. Rev Assoc Med Bras 2010; 56: 655-659. 5. Elsheikh M, Dunger DB, Conway GS, Wass JA: Turner’s Syndrome in Adulthood. Endocrine Reviews 2002; 23: 120-140. 6. Volkl T, Degegenhardt K, Skoch A et al.: Cardiovascular Anomalies in Children and Young Adults with Ullrich-Turner Syndrome – The Erlangen Experience. Clin Cardiol 2005; 28: 88-92. 7. Sybert V: Cardiovascular Malformations and Complications in Turner Syndrome. Pediatrics 1998; 101: 1-7. 8. Loscalzo ML, Van PL, Ho VB et al.: Association between fetal lymphedema and congenital cardiovascular defects in Turner syndrome. Pediatrics 2005; 115: 732-735. 9. Lin A, Lippe B, Rosenfeld R: Further Delineation of Aortic Dilation, Dissection, and Rupture in Patients With Turner Syndrome. Pediatrics 1998; 102: 1-9. 10. Gravholt CH, Landin-Wilhelmsen K, Stochholm K et al.: Clinical and epidemiological description of aortic dissection in Turner’s syndrome. Cardiol Young 2006; 16: 430-436. 11. Elsheikh M, Casadei B, Conway GS, Wass JA: Hypertension is a major risk factor for aortic root dilatation in women with Turner’s syndrome. Clin Endocrinol 2001; 54: 69-73. 12. Gravholta C, Wurgler K, Erlandsenc M et al.: Nocturnal hypertension and impaired sympathovagal tone in Turner syndrome. Journal of Hypertension 2006; 24: 353-360. 13. Carel JC, Mathivon L, Gendrel C et al.: Near normalization of final height with adapted doses of growth hormone in Turner’s syndrome. J Clin Endocrinol Metab 1998; 83: 1462-1466. 14. Rongen-Westerlaken C, Corel L, van den Broeck J et al.: Reference values for height, height velocity and weight in Turner’s syndrome. Swedish Study Group for GH treatment. JM Acta Paediatr 1997; 86: 937-942.
